In Chap. 6 we introduced how to cluster sequences into OTUs using closed-reference clustering, de novo clustering, and open-reference clustering. In this chapter we first briefly introduce the history of numerical taxonomy (Sect. 7.1). Then we introduce the principles and aims of numerical taxonomy (Sect. 7.2). Next, we briefly describe the philosophy of numerical taxonomy (Sect. 7.3). Followed that in Sect. 7.4, we focus on the formation and characteristics of commonly clustering-based OTU methods, which have a large impact on microbiome study in both concept and methodology. In Sect. 7.5, we discuss statistical hypothesis testing of OTUs. Section 7.6 describes the characteristics of clustering-based OTU methods. Finally, we conclude this chapter with a summary (Sect. 7.7).
In general, all the methods used in numerical taxonomy can be called numerical taxonomic methods. Here, we define the methodology of numerical taxonomy in the non-rigorous way and name the methods used in numerical taxonomy as “OUT-based methods” or “OTU methods,” which include defining OTUs, measuring taxonomic resemblance (similarity/dissimilarity), clustering-based OTU methods, factor analysis, ordination, and discriminant function analysis because in numerical taxonomy all the used methods mainly regard construction of taxonomic structure by classifying and identifying OTUs. We specifically refer to all the clustering methods used in numerical taxonomy as clustering-based OTU methods.
Numerical taxonomy has directly affected several research fields, e.g., phylogenetics, genetics, and paleontology (Sokal and Sneath 1963), especially ecology, psychometrics (Sneath 1995), and microbiome. The clustering-based OTU methods in microbiome studies were originated in OTU methods in numerical taxonomy founded in 1963 by microbiologist Peter H. A. Sneath and population geneticist Robert R. Sokal. The lineage between the fields of numerical ecology and numerical taxonomy had been described in (Legendre 2019).
7.1 Brief History of Numerical Taxonomy
Michener and Sokal (Michener and Sokal 1957; Sokal 1958a) studied coding and scaling multistate characters, parallelism and convergence, and equal weighting of characters. Sokal’s work (Sokal 1985) originated from how to efficiently classify group of organisms and contributed his biological statistics. He developed the average-linkage clustering methods, which were inspired by various types of primitive cluster and factor analyses in the 1940s by Holzinger and Harman (1941), Cattell (1944), Tryon’s cluster analysis (Tryon 1934), and as well Thurstone on factor analysis (Vernon 1988).
Sneath (Sneath 1957a, b) presented his principles and methodology, which were built on a similarity coefficient and Jaccard’s coefficient and single-linkage clustering (Sokal 1985).
Cain and Harrison (1958) were the first to differentiate clearly phenetic from cladistic relationships (Sokal 1985; Sneath 1995) and to apply average distance to numerical taxonomy (Sokal 1985). Cain’s two other papers (Cain 1958, 1959) also contributed to the early establishment of numerical taxonomy (Sneath 1995) in which he discusses the logic and memory in Linnaeus’s system of taxonomy and the deductive and inductive methods in post-Linnaean taxonomy, respectively.
Rogers and Tanimoto (1960) developed the early methods of probabilistic distance coefficients and clustering numerical taxonomy (Sokal 1985; Sneath 1995). George Estabrook, a distinguished student from this group, also made contributions to numerical taxonomy and especially made outstanding contributions to numerical phylogenetic inference (Sokal 1985; Sneath 1995).
Gilmour (1937, 1940, 1951) who provided some key concepts of information content and predictivity that they adapted from the philosophies of science of Mill (1886) and Whewell (1840).
Simpson’s clear thought on systematics and the problems confronting them had major influence on Sokal and Sneath and contributed to the founding of numerical taxonomy, although not from methodology (Simpson 1944, 1961).
Previously, the methods for quantifying the classificatory process in systematics had been called “quantitative systematics.” In 1960, Sokal and Sneath renamed these methods collectively as “numerischen taxonomie” (“numerical taxonomy”) (Sokal 1960, 1961; Rohlf and Sokal 1962) (On September 15, 1982, Sokal said in fact they coined the name “numerical taxonomy,” This Week’s Citation Classic, CC/Number 46, November 15, 1982). Sokal and Sneath in particular became acknowledged fathers of numerical taxonomy (Vernon 1988; Goodfellow et al. 1985). For the detail founding history of numerical taxonomy, we reference the interested readers to the review articles by Sokal (1985), Vernon (1988), and Sneath (1995) as well as references therein.
First methods and theory of numerical taxonomy were developed during 1957–1961 (Sneath and Sokal 1973), while in 1962, Rohlf (1962) first conducted hypothesis test of the nonspecificity; Sokal and Rohlf (1962) first used criterion of goodness of a classification to compare dendrograms in numerical taxonomic studies. In 1962, when the book Principles of Numerical Taxonomy (Sokal and Sneath 1963) was still in preparation, Sneath and Sokal published an article called “Numerical Taxonomy” in Nature (Sneath and Sokal 1962). In this article, the comprehensive theory and methods of numerical taxonomy were first published. In this Nature article, Sneath and Sokal defined their named field “Numerical Taxonomy” as “the numerical evaluation of the affinity or similarity between taxonomic units and the ordering of these units into taxa on the basis of their affinities” (Sneath and Sokal 1962, p. 48). “The term may include the drawing of phylogenetic inferences from the data by statistical or other mathematical methods to the extent to which this is possible. These methods will almost always involve the conversion of information about taxonomic entities into numerical quantities” (Sokal and Sneath 1963, p. 48). In their 1973 book, the term “numerical taxonomy” (Sneath and Sokal 1973; Sokal 1985) was clearly defined as “the grouping by numerical methods of taxonomic units into taxa on the basis of their character states. The term includes the drawing of phylogenetic inferences from the data by statistical or other mathematical methods to the extent to which this is possible. These methods require the conversion of information about taxonomic entities into numerical quantities” (Sneath and Sokal 1973, p. 4).
7.2 Principles of Numerical Taxonomy
Numerical taxonomy is an empirical science on a classification study and a multivariate technique focusing on problems of classification and based on multiple characters. The basic ideas of numerical taxonomy were to use large numbers of unweighted characters through statistical techniques to estimate similarity between organisms. In other words, numerical taxonomy is based on shared characters and by clustering their similarity values to create groups and to construct the classifications without using any phylogenetic information (Sokal 1963). It aims to (1) obtain repeatability and objectivity [see (Sokal 1963, pp. 49–50; Sneath and Sokal 1973, p. 11)]; (2) quantitatively measure resemblance from numerous equally weighted characters (Sokal 1963, pp. 49–50); (3) construct taxa from character correlations leading to groups of high information content (Sokal 1963); and (4) separate phenetic and phylogenetic considerations (Sokal 1963).
Numerical taxonomy has been built on seven fundamental principles (Sneath and Sokal 1973, p. 5): “(1) The greater the content of information in the taxa of a classification and the more characters on which it is based, the better a given classification will be. (2) A priori, every character is of equal weight in creating natural taxa. (3) Overall similarity between any two entities is a function of their individual similarities in each of the many characters in which they are being compared. (4) Distinct taxa can be recognized because correlations of characters differ in the groups of organisms under study. (5) Phylogenetic inferences can be made from the taxonomic structures of a group and from character correlations, given certain assumptions about evolutionary pathways and mechanisms. (6) Taxonomy is viewed and practiced as an empirical science. And (7) Classifications are based on phenetic similarity.”
Based on the 7 principles, Sokal and Sneath criticized that the practices of previous taxonomy may yield arbitray and artificail taxa (Sokal 1963, p. 9) because previous taxonomy recognized and defined taxa were based on three often not clearly separated kinds of evidence (Sokal 1963, p. 7): (1) Resemblances (a taxon is formed based on phonetically more resemble entities), (2) Homologous characters (a taxon is formed by entities sharing characters of common origin), and (3) Common line of descent (a taxon is formed with membership in a common line of descent). The evidence of type 3 is rare and hence taxa are usually inferred from types 1 and 2, while the conclusions based on homologies (evidence of type 2) are often deducted from phylogenetic speculations (evidence of type 3).
Different from the traditional subjective approach, the approach of numerical taxonomy is to classify organisms by statistical procedures (Vernon 1988). The underlying reason to propose a new field of taxonomy was a dissatisfaction with the evolutionary basis of taxonomy (Vernon 1988) or a major concern of producing a classification in accordance with phylogeny. In other words, for numerical taxonomy, relationships must be determined in a non-historical sense and only then the most likely lines of descent can be decided on it (Michener and Sokal 1957).
The principles or framework of numerical taxonomy consists of several core ideas and statistical procedures. The main point is how to construct a taxonomic system. Understanding these principles will facilitate better understanding of numerical taxonomy, why the methodology of numerical taxonomy has been adopted into microbiome study, as well as the current challenges using the methodology in microbiome study. Construction of a taxonomic system is to find taxonomic evidence, which is relevant to defining characters and taxa.
In this section, we review how numerical taxonomy defines characters and taxa (the fundamental taxonomic units) as well as obtains estimations of affinity between taxa from the data (characters).
7.2.1 Definitions of Characters and Taxa
Numerical taxonomy defines characters entirely based on the differences between individuals [see (Sokal and Sneath 1963, p. 62; Sneath and Sokal 1973, p. 72)]; thus a character is a property or “feature which varies from one kind of organism to another” (Michener and Sokal 1957) or anything that can be considered as a variable logically (rather than functionally or mathematically) independent of any other thing (Cain and Harrison 1958).
Unit characters are defined in terms of information theory [see (Sokal and Sneath 1963, p. 63; Sneath and Sokal 1973, p. 72)], which is to allow unit characters to be treated in a broad way, that is, to convey information. Thus, a unit character (or a “feature”) is an attribute possessed by an organism for yielding a single piece of information.
During 1960s–early 1970s, in most cases it is difficult to make genetic inferences from phenetic studies of characters. The considerations of defining unit characters in terms of information theory were premature; thus, Sokal and Sneath proposed a narrow working definition of unit character (phenotypic characters). A unit character is defined as taxonomic character of two or more states, which within the study at hand cannot be subdivided logically, except for subdivision brought about by changes in the method of coding (Sokal and Sneath 1963, p. 65). Sokal and Sneath roughly grouped taxonomic characters into four categories [see Sokal and Sneath (Sokal and Sneath 1963, pp. 93–95; Sneath and Sokal 1973, pp. 90–91)]: (1) morphological characters (external, internal, microscopic, including cytological and developmental characters); (2) physiological and chemical characters; (3) behavioral characters; and (4) ecological and distributional characters (habitats, food, hosts, parasites, population dynamics, geographical distribution).
One important impact of defining unit character in terms of information lies on the factor that many bits of information contained in the simplest organisms can be interpreted in terms of molecular genetics.
Among the taxonomic characters the chemical characters are very important. They are the nucleotide bases of the nucleic acids (often the DNA of the genome) in their specific sequences (Sneath and Sokal 1973, p. 94). These characters are created by the chemical methods: the nucleic acid pairing technique which was pioneered by Doty et al. (1960), and advanced particularly by McCarthy and Bolton (1963). The characters of nucleotide bases of the nucleic acids are closer to the genotype, and the chemical data can be arranged in the order, and hence their superiority over to morphological characters have been discussed among biochemists; but at that time (1973) numerical taxonomy was not optimistic on these characters in their practical applications (Sneath and Sokal 1973, p. 94). The study of DNA sequences in numerical taxonomy has not largely been explored. But the problems or challenges of how to define these kinds of characters and more precisely and profoundly analyze the nature of phenetic similarity have been described (Sneath and Sokal 1973).
Actually, the microbiome study is just based on these characters. The later microbiome sequecing study originated in coding genetic information and defining unit character in terms of information.
Sokal and Sneath suggested all known characters should be included to avoid bias (or spurious resemblance between characters). They also suggested that each character should be given equal weight because no method can satisfactorily allocate weights on characters.
Sokal and Sneath adopted Gilmour naturalness as a criterion for natural classifications in phenetics and thought that most taxa in nature (not only biological taxa) are polythetic. The term “nature” is used in the sense implied that suggests a true reflection of the arrangement of the organisms in nature. For them, no single state is either essential or sufficient to make an organism a member of the group; in a polythetic taxon membership is based on the greatest number of shared character states. Sokal and Sneath defined “natural” taxa as the particular groups of organisms that are studying. In other words, natural taxa are based on the concept of “affinity,” which is measured by taking all characters into consideration and the taxa are separated from each other by means of correlated features (Sokal and Sneath 1963, p. 16). Thus, naturalness is a matter of degree rather than an absolute criterion of a classification (Sokal 1986). Depending on the number of shared character states, different classifications will consequently be more or less natural.
7.2.2 Sample Size Calculation: How Many Characters?
First, we need to point out that numerical taxonomy uses the notations of sample size and variable in a different way from many multivariate statistics. In multivariate statistics, n is used for sample size, and p for variable number. However, in numerical taxonomy, sample size represents the number of OTUs (denoted as t); character represents the variable (the variable number is denoted as n). In microbiome study, we can consider n as sample size and p as variable number in an OTU table that is originated in numerical taxonomy. In later numerical ecology, similar usages of variable and object have been adapted. In numerical ecology, the terms descriptor and variable are used interchangeably to refer to the attributes, characters, or features for describing or comparing the objects of the study, while the term objects is used to refer to the sites, quadrats, observations, sampling units, individual organisms, or subjects (Legendre and Legendre 2012, p. 33). Legendre and Legendre (2012) have point out that ecologists also called observation units as “samples”; and they used the term sample in its statistical sense, which refers to a set of sampling observations. They called individuals or OTUs (operational taxonomic units) used in numerical taxonomy as objects (p. 33). This usage is consistent with that of numerical taxonomy. However, in microbiome study, the terms sample and variable are used in the different way from numerical taxonomy. In microbiome study, both terms of sample and variable are used in their statistical senses, in which samples are used to refer to observation units, whereas OTUs are used as variables. Thus, n is used for sample size, and p or m for variable number or how many OTUs or taxa. Therefore, when we talk about the statistical issues such as large p and small n problems, we have used them in this way (i.e., in most multivariate statistics) [See (Xia et al. 2018; Xia 2020; Xia and Sun 2022) and also Chap. 18 in this book]. Another different use in numerical taxonomy is R-and Q-Techniques (see Sect. 7.4.2.2 for details).
Power and sample size calculations are challenging in experimental studies and clinical trials, especially for microbiome related projects. It is very challenging to estimate the power and sample size in numerical taxonomy and microbiome studies. At the beginning when numerical taxonomy was established, Sokal and Sneath recognized that it was very complicated to obtain the required number of characters to estimate the similarity with given confidence interval and significance of probability level (Sokal and Sneath 1963; Sneath and Sokal 1973).
Generally, the number of characters chosen to estimate of an overall similarity is based on the matches asymptote hypothesis and an empirical observation: a given estimate of phenetic similarity is favored with much different phenetic information provided by large number of characters. Overall, numerical taxonomy is a large sample method; as a general rule, it emphasizes to use more rather than fewer characters. To achieve overall predictivity classification and overall phenetic similarity, taxa should be based on many characters.
- (1)
The reliability of the estimates of 95% confidence limits of the correlation coefficients (Sokal and Sneath 1963, pp. 115–116). The algorithm is that as sample size (number of characters) increases, the confidence limits of the similarity coefficients will be narrowed. Thus, if the confidence limits will be getting sufficiently narrow, then sampling error is not likely to be mistaken for real differences in relationship. Based on this criterion, Michener and Sokal (1957) suggested at least sixty characters are needed to give sufficiently narrow confidence limits. This criterion is obviously arbitrary.
- (2)
A sampling strategy of phenotypic characters based on molecular considerations. This strategy considers each character as a sample of the genome and determines when a required percentage of the genome has been sampled at a required confidence level. However, since any reasonable number of characters represents a relatively small percentage of the genome, this approach is premature and the links between the molecular basis of the genome and the phenetic characters are limited (Sneath 1995). Thus, this approach cannot be used to calculate sample size (Sokal 1963, p. 117).
- (3)
An empirical evidence-based numerical classification. Sokal and Rohlf (1970) found how many independent individuals coding can describe the same variation patterns and additional independently varying characters would not substantially change the classification or the underlying character-correlational structure. Later, Sneath and Sokal (1973, p. 108) suggested performing a factor analysis to check how many reasonable number of characters is employed to stabilize common factors (taxonomic structure).
- (4)
A simulation experiment was suggested to test the different correlational structure to see the effect of the addition of new characters, that is, to check how many reasonable numbers of characters is employed to stabilize the matrix correlation of successive similarity matrices. Two distinct models or distribution of characters (J-shaped curve similar to a Poisson distribution with a low mean and uniform distribution) were also used in this verification of the asymptotic approach to taxonomic stability (Sneath and Sokal 1973, p. 108).
This large sampling theory and experience-based sample size calculation and power analysis are continued to be used in microbiome study. However, these approaches do not often lead to a satisfactory result. Sokal (1985) acknowledged that the genetically based estimate of an overall similarity based on the matches asymptote hypothesis of Sokal and Sneath (1963) to choose the characters is not useful to solve the increasing complex relations underlying molecular genetics. Sample size calculation and power analysis are especially challenging in microbiome research, because in a microbiome study, we not only need to determine how many samples are required to obtain sufficient power in classification of OTUs, but also need to determine how many samples in statistical analysis could lead to a robust result.
On one hand we really do not know or rarely considered how many samples that are needed to process sequencing to obtain OTUs or ASVs; on the other hand, we also do not know for sure how many samples are required for downstream statistical analysis to obtain sufficient power to detect the hypothesis testing of outcome. Currently, the most proposed methods in microbiome study are based on simulation and use the detected effect sizes in a given database or study as the input sample size.
7.2.3 Equal Weighting Characters
For Sokal and Sneath, “weighting” (Sneath and Sokal 1973, p. 110) or a priori weighting (Sneath and Sokal 1973, p. 109) is for general taxonomic analyses or general nature of natural classifications and is commenced before a classification. They advocated to give each character (feature) equal weight in the computation of a resemblance coefficient or in creating taxonomic groups (natural taxa). Equal weighting is based on seven reasons (Sneath and Sokal 1973, pp. 112–113): (1) we cannot decide how to weight the features, (2) we cannot prejudge the importance of characters, (3) the concept of taxonomic importance has no exact meaning, (4) we cannot give exact rules for differential weighting, (5) the nature of a taxonomy is for general purposes, and it is inappropriate to give greater weight to features for general use, (6) the property of “naturalness” lies on its high implied information by a natural group, which is irrelevant to its importance, and (7) the use of many characters makes the effective weight to be clear that each character contributes to the similarity coefficients.
We should point out, at the beginning, Sokal and Sneath emphasized the importance of equal weighting of characters and the impossibility of unequal weighting. However, to respond to the weighting approaches proposed in numerical taxonomy literature, Sokal (1986) recognized, in practice, strictly equal weighting of characters is impossible because the omitted characters have zero weight, and the similarity is mainly determined by the characters with many states than those with few states. Particularly, in their 1973 book, they stated that unequal weighting in phenetic taxonomy can be carried out by an explicit algorithm, such as the rarity of features or their commonness in the whole set of OTUs can be used to weight the characters (Sneath and Sokal 1973, p. 111). However, they thought that the results using unequal weighting likely will be little different from those using equal weighting and most work in phenetic numerical taxonomy has avoided explicit weighting because no algorithm is convincing for doing so.
For the detail discussions on weighting, the interested reader is referred to (Sokal 1963, pp. 50–51; pp. 118–120; Sneath 1957b; Michener and Sokal 1957; Sneath and Sokal 1973, pp. 109–113).
7.2.4 Taxonomic Rank
Generally three criteria for taxonomic rank in numerical taxonomy have been applied (Sneath and Sokal 1973, p. 60): (1) Both phenetic and genetic criteria are commonly used although for biological species and below level, these criteria may be in conflict. (2) In the absence of data on breeding and in all apomictic groups (which is the most major cases in systematics), the phenetic similarity between the individuals and on phenotypic gaps are used to define species. (3) The higher rank of categories is per forcedly defined using the phenetic criteria.
In numerical taxonomy, the species are based on the phenetic similarity between the individuals and on phenetic gaps. The taxonomic rank is in the sense of phenetic rank, which is based on affinity alone (Sokal and Sneath 1963, pp. 30–31). However, since the end of 1960s, numerical taxonomists has recognized that there are no criteria for any absolute measure of taxonomic rank, and criteria of phenetic rank is difficult to apply to all fields (Sneath and Sokal 1973, p. 61). For example, in protein and DNA, it was shown that widely different phenetic differences exist at the same taxonomic ranks. It was difficult to use the age of clades to measure the taxonomic rank. Overall, Sneath and Sokal noticed that it was difficult to define the taxa of low rank and to arrange them hierarchically (Sneath and Sokal 1973, p. 62).
In summary, Sneath and Sokal (1973, p. 290) believed that in practice taxonomists should usually use phenetic criteria to measure rank and that the most satisfactory criterion of a rank of a single taxon is some measure of its diversity. Of course, the criteria of taxonomic rank are challenging and debatable not only in numerical taxonomy but also in microbiome study.
7.3 Phenetic Taxonomy: Philosophy of Numerical Taxonomy
Here, to better understand phenetic taxonomy, and related methods that have been adopted into microbiome study, we describe the standing points of philosophy of numerical taxonomy. We should distinguish phenetic taxonomy (or phenetics) from phenetic techniques in numerical phenetics. The former is a theory or philosophy or of classification, and the latter are the methods developed to apply the theory.
Three common taxonomic philosophies exist in literature: phenetic philosophy, phylogenetic (cladistic) philosophy, and evolutionary philosophy. Overall, the philosophy of numerical taxonomy is the movement from the metaphysics of essentialism to the science of experience, from deductive methods to inclusive methods, from evolutional phylogenetics to numerical phenetics based on similarity.
7.3.1 Phenetics: Numerical-Based Empiricism Versus Aristotle’s Essentialism
Some descriptions of Aristotle’s metaphysics of essentialism (metaphysical essentialism) and Darwin’ theory of evolution will provide a basic background to understand numerical phenetics.
7.3.1.1 Aristotle’s Metaphysics of Essentialism
Essentialists believe true essences exist. Aristotle defined essence as “the what it was to be” for a thing (Aristotle 2016) or sometimes used the shorter term “the what it is” for approximating the same idea. Aristotle defined essence as analyzed entities. For example, in his logical works (Aristotle 2012), Aristotle links essence to definition—“a definition is an account (logos) that signifies an essence” or “there will be an essence only of those things whose logos is a definition,” “the essence of each thing is what it is said to be intrinsically.” Aristotle’s essence is related to a thing. In his classic definition of essence, a thing’s essence is that which it is said to be per se, or is the property without which the thing would not exist as itself. Therefore, essence is constitutive of a thing, which is eternal, universal, most irreducible, unchanging. The classical example of essence definition is that Aristotle defined the human essence: to be human is to be a “rational animal.” The “rational” is the species or specific difference, which separates humans from everything else in the class of animals.
Another important essentialist concept is forms. For Plato, forms are abstract objects, existing completely outside space and time. Aristotle criticized and rejected Plato’s theory of forms but not the notion of form itself. For Aristotle, forms inform the thing’s particular matter. Thus, forms do not exist independently of things—every form is the form of some things. He called “the form” of a particular thing (Albritton 1957).
The naturalists in seventeenth and eighteenth centuries who attempted to classify living organisms were actually inspired by the terms and definitions from the Aristotelian system of logic. Darwin traced evolutionary ideas (the principle of natural selection) back to Aristotle in his later editions of the book of The Origin of Species (Darwin 1872), and commented on Aristotle on forms.
Aristotelian system attempted to discover and define the essence of a taxonomic group (“real nature” or “what makes the thing what it is”). Aristotelian logic is a system of analyzed entities rather than a system of unanalyzed entities.
7.3.1.2 Natural Classification: Numerical-Based Empiricism
Numerical taxonomy is a largely empirical science (Sokal 1985; Sneath 1995).
Natural classification was developed based on the philosophy of empiricism and was affected by the philosophy of the inductive sciences. Some key concepts of information content and predictivity provided by John Gilmour (Sneath 1995) who was one of the early contributors to the founding of numerical taxonomy were adapted from the Victorian philosophers of science John Stuart Mill (1886) and William Whewell (1840).
Sokal and Sneath criticized that Aristotelian logic cannot lead to biological taxonomy because the properties of taxonomy cannot be inferred from the definitions [see (Sokal and Sneath 1963, p. 12; Sokal 1962; Sneath and Sokal 1973, p. 20)]. Thus, they emphasized that natural taxa do not necessarily possess any single specified feature, which is the Aristotelian concept of an essence of a taxon, because natural or “general” classification or natural groups are in logic unanalyzed entities (Sokal and Sneath 1963, pp. 19–20).
Sneath and Sneath (1962) differentiated the terms “polythetic” and “monothetic” in taxonomic groups. The monothetic groups define a unique set of features that is both sufficient and necessary for membership in the group thus defined. The monothetic groups have the risk to misclassify natural phenetic groups and hence do not yield “natural” taxa. In contrast, in a polythetic group, organisms that have the greatest number of shared character states are placed together, and hence no single state is either essential to group membership or is sufficient to make an organism a member of the group.
For Sneath and Sokal, natural taxa are usually not fully polythetic, and it is possible that they are never fully polythetic; however, in practice, they suggest considering the possibility of a taxon being fully polythetic due to limited knowledge of whether any characters are common to all members (Sneath and Sokal 1973, pp. 20–21).
Numerical taxonomists emphasize that biological taxonomy is a system of unanalyzed entities. Their properties cannot be inferred from the definitions, instead through classification. In his “Taxonomy and Philosophy” (Gilmour 1940), Gilmour stated that classification enables us to make generalizations and predictions. The more information obtained by classification enables us to make more predictions. The more predictions the classification is more “natural.”
For Gilmour, the most “natural” system/classification contains the most information, which is highly predictive and serves the most general purpose (Vernon 1988), while less “natural” classification is very useful for specific purposes. Generally Sokal and Sneath held with Gilmour’s review of “natural” classification. However, classification serves as special purpose. Such classification is a special classification and is often “arbitrary.” Thus Gilmour’s classification conveys less information than a general or “natural” one. For Sokal and Sneath, a natural taxonomic group or natural taxa of any rank can be classified. For example, the genetic natural classification can be supported (Vernon 1988). They defined a natural classification in phenetics as one in which the members of each taxon at each level are on the average more similar to each other than they are to members of other taxa at corresponding levels (Sokal 1986).
The philosophical attitude of numerical taxonomy in systematics is empiricism and consequently is not committed to the existence of biological species (Sokal and Crovello 1970). Put together, numerical taxonomy is both operational and empirical. Numerical-based natural classification is an empiricism. Empiricism in taxonomy is the only reasonable approach for arranging organized nature (Sokal 1963, p. 55). Sneath and Sokal (1973, p. 362) emphasized that numerical taxonomy is an empiricism and operationism and thought that the biological species definition was difficult to apply and in practice was nonoperational.
7.3.2 Classification: Inductive Theory Versus Darwin’ Theory of Evolution
Sneath (1995) considered Sokal’s and his numerical taxonomy as the greatest advance in systematics since Darwin or even since Linnaeus because Darwin had relatively little effect on taxonomic practice (Hull 1988). This statement emphasizes that the numerical taxonomy has the different taxonomic methodology from Darwin’s taxonomic method.
Darwin in 1859 published his On the Origin of Species (or, more completely, On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life) (Charles Darwin 1859), which is considered to be the foundation of evolutionary biology. Darwin’s scientific theory introduced in this book stated that populations evolve over the course of generations through a process of natural selection. In the 1930s and 1940s, the modern evolutionary synthesis had integrated Darwin’s concept of evolutionary adaptation through natural selection with Gregor Mendel’s theories of genetic inheritance (McBride et al. 2009) to become a central concept of modern evolutionary theory and even unifying concept of the life sciences.
The arguments of Darwin’s theory of evolution are combined with facts/observations and the inferences/speculations drawn from them (Mayr 1982). His theory of classification stated in Chapter XIII of his book on the origin of species (Darwin 1859) starts by observing that classification is more than mere resemblance, and it includes propinquity of descent (the only known cause of the similarity of organic beings) which is bonded and hidden by various degrees of modification (pp. 413–414). For Darwin, the natural system is founded on descent with modification and the characters that have been shown having true affinity between any two or more species, are those which have been inherited from a common parent, and, in so far, “all true classification is genealogical” (Darwin 1859, p. 420).
Obviously above Darwin’s theory of classification is deductive although he said later that organic beings can be classed in many ways, either artificially by single characters or more naturally by a number of characters. Thus classification is not related to genealogical succession (Darwin 1886, p. 364). Darwin’s work contributed to the recognition of species as real entities (Sokal and Crovello 1970).
The influence of Darwin’s theory of evolution lies on the fact that systematics was considered as phylogenetically, in which a taxon was interpreted as a monophyletic array of related forms. However, the change of philosophy was not accompanied with a change in method (Sokal 1963, p. 20). Thus, during the early 1950s, some taxonomists began to suspect the appropriateness of the evolutionary approach to classification in phylogenetic taxonomy and reexamined the process of classification. For example, Mayr and Simpson recommended highly weighting those characters which indicated evolutionary relationships. However, for those characters with the absence of fossils and knowledge of breeding patterns, this was very challenging and thus the phylogenetic importance of characters could be given due to much speculation.
The trend to reject phylogenetic speculation in classification has been reviewed as background that Sokal and Michener, Cain and Harrison, and Sneath developed their ideas of the numerical taxonomy (Vernon 1988). Under the influence of the Darwinian theory of evolution, the natural classifications of earlier taxonomists can be explained and understood in terms of descent from a common ancestor. That is, they now interpreted a taxon as a monophyletic array of related forms (Sneath and Sokal 1973, p. 40). However, the change of the Darwinian philosophy did not bring a methodology change in taxonomy. It only changed its terminology.
Essentially the methodology of the theory of classification in numerical taxonomy is inductive, and hence it was arising against Darwinism’s deductive theory of classification. Sokal and Sneath (1963, p. 21) criticized that the phylogenetic approach cannot be used to establish natural taxa because it lacks the fossil record. They emphasized that classification cannot describe both affinity and descent in a single scheme [see (Michener and Sokal 1957; Sokal 1963, p. 27)]. For them, deductive reasoning in tracing phylogenies is dangerous (Sokal 1963, p. 29).
7.3.3 Biological Classifications: Phenetic Approach Versus Cladistic Approach
As a field, numerical taxonomy provides explicit principles of phenetics through numerical approaches to phylogeny and emphasizes the distinction between phenetic and cladistic relationships (Sneath 1995), which was first clearly stated by Cain and Harrison (1960). Numerical taxonomy has a broader scope than phenetics. It includes both phenetic and phylogenetic approaches. It includes the drawing of phylogenetic inferences from the data by statistical or other mathematic methods to the extent that this was possible [see (Sokal 1963, p. 48; Sneath 1995)].
A phenetic classification attempts to establish taxa either based on maximum similarity among the OTUs or on maximum predictive value (homogeneity of character states) (Sokal 1986). This approach assumes but never rigorously demonstrated (Farris 1979) that high similarity among individuals in a taxon would result in high predictive values for many of the characters.
For an approach of phenetic phylogenetic classifications, most phenetic methods do not attempt to maximize similarities in taxa but settle for proven suboptimal solutions such as average-linkage clustering (Sokal 1986). However, once a phenetic classification has been established, it is possible to attempt to arrive the phylogenetic evidence and phylogenetic deductions.
Sneath and Sokal (1973) divided and refined phylogenetic relationships into phenetic and cladistic relationships (p. 41). Sokal and Sneath clearly made distinction between phenetic and cladistic classifications (Sokal 1985) and considered it as an important advance in taxonomic thinking for separation of overall similarity (phenetics) from evolutionary branching sequences (cladistics) (Sneath and Sokal 1973, p. 10). Thus, the phylogeny that the numerical taxonomy aimed to establish is first to make the distinction between phenetic and cladistic relationships (Sneath 1995).
Natural taxonomic groups have the evolutionary basis, and the observed phenetic diversity of living creatures is a reflection of evolution (Sokal 1963). In other words, natural taxonomic groups just reflect the restriction of evolution and the accumulation with the time genetic difference.
Phylogeny or cladistic relationships cannot be used to construct phyletic classification. Numerical classification principally is a natural classification, which is to be natural rather than artificial. It truly reflects the arrangement of the organisms in nature. Generally natural taxa are considered to be those monophyletic taxa that reflected phylogenetic history.
Even if phylogeny could be used to create a valid phylogenetic classification, it is not necessarily desirable. Phyletic classifications could be chaotic in classification of viruses and especially in bacteriophages, and phenetic classification is the only way to classify them. And the validity of a phylogenetic classification may be in a serious doubt.
Overall phenetic similarities between organisms can be used to make cladistic deductions. Phenetic clusters based on living organisms are more likely to be monophyletic; in the absence of direct evidence, phenetic resemblance is the best indication of cladistic relationships.
A phenetic approach should be chosen to classify natural taxa. Numerical taxonomy uses phenetic evidence to establish a classification. Although both phenetic and cladistic approaches have problems to the study of taxonomic relationships, the phenetic approach is the better choice to be used in classifying nature. First, the problems of estimating phenetic relationships are largely technical challenges and can be overcome, whereas the problems of estimating cladistic sequences are difficult to be solved. Especially numerical cladistics cannot provide necessarily cladistic sequences. Second, since in the fossil record only very small portion of phylogenetic history is known, in the vast majority of cases phylogenies are unknown and possibly unknowable; it is difficult to use the phylogenetic approach in systematics/taxonomy to make use of phylogeny for classification. Thus, it is almost impossible to establish natural taxa based on phylogenies. In contrast, a phenetic classification is at least a self-sufficient, factual procedure and hence can provide the best classification in most cases although it cannot explain all biological phenomena.
In general, a phenetic classification is superior to a cladistic classification from two viewpoints: domain and function.
It is difficult to define or obtain clear decisions on monophyly. Taxa in orthdox taxonomy should be in phenetic groups rather than in general monophyletic groups (clades). There are three reasons: (1) Phenetic and phyletic relations are two formally independent taxonomic dimensions. (2) It is expected that the phenetic groups in great majority of examples will indeed be clades based on the assumption that close phenetic similiarity is usually due to close relationship by ancestry. (3) In general classfication, phenetic taxa is perfered to cladal taxa, while employing clades has the advantages only in the study of evolution.
Phenetics aims to give information-rich groups, while the goal of phylogenetics is to reconstruct evolutionary history. Phenetic groupings can be verified by phenetic criteria but cannot be proven to correspond to reality, whereas phylogenetic groupings must correspond to reality, but cannot be verified (Sneath 1995). The reason is that information-rich groups do not necessarily have a historical basis. Actually, the theories of phenetics involve three types of theory (Sneath 1995): (1) theory of homology (i.e., what should be compared with what), (2) theory of information-rich groupings (which implies high predictivity) (Mill 1886; Gilmour 1940), and (3) Mill’s (1886) theory of general causes (i.e., phenetic groups are due to as yet undiscovered causes, which linked numerical taxonomy more closely to Darwin’s the origin of species) (Sneath 1995).
We should point out, at the beginning, numerical taxonomy still attempted to establish phylogenetic classifications (Michener and Sokal 1957). However, subsequently, the numerical taxonomists realized that because different lineages may evolve at different rates, it is impossible to draw a phyletic tree in which all the lineages evolve at the same rate and or to derive evolutionary rates from similarity coefficients among recent forms. Thus, they claimed that phenetic classification is the only consistent and objective classification (Sokal and Sneath 1963, pp. 229–230).
In summary, Sokal and Sneath agreed with Bigelow (1961) that a horizontal (phenetic) classification is more satisfactory than a vertical (phyletic) classification (Sokal and Sneath 1963, p. 104). In terms of vertical classification and horizontal classification (Simpson 1945), numerical taxonomy groups taxa based on horizontal classification. However, in numerical taxonomy, a phenetic taxonomic group does not need to be necessarily identical with a phyletic group (Sokal and Sneath 1963, p. 32).
7.4 Construction of Taxonomic Structure
Phenetic taxonomy (classification) aims to arrange operational taxonomic units (OTUs) into stable and convenient classification (Sokal 1986), which is to group the most similar OTUs. Thus, various statistical methods that serve the goals of grouping OTUs used in numerical taxonomy can be called OTU-based methods. Among these methods, the concept of OTUs, clustering analysis methods, the coefficients of similarity, distance, association, correlation, and ordination techniques have the most impacts on microbiome study. Actually, ordination techniques have been most fully developed in ecology. The ordination methods used in later numerical taxonomy were adopted or influenced by ecology. We comprehensively investigate ordination methods in Chap. 10. In this section, we focus on the review of the concept of OTUs, clustering analysis methods, the coefficients of similarity, distance, association, and correlation.
The empirical inclusive methodology of numerical taxonomy has impacted many other research fields. Numerical taxonomy has expanded into ecology and psychometrics at the later 1970s and early 1980s. The numerous applications in ecology were summarized by Legendre and Legendre (1983; Sneath 1995). The methodology of numerical taxonomy and especially the clustering methods have been adopted in numerical ecology and later microbiome fields. For example, single-linkage, complete-linkage, and average-linkage clustering methods were adopted and especially the average-linkage clustering method was advocated by the microbiome software mothur.
Various clustering analysis methods were developed and employed to measure homogeneity of OTUs regarding character states to maximize homogeneity (Sneath and Sokal 1973; Sokal 1985). We term these methods that were adopted from numerical taxonomy as the commonly clustering-based OTU methods. Here we characterize them as below.
7.4.1 Defining the Operational Taxonomic Units
The taxonomic units that numerical taxonomy classifies are not the fundamental taxonomic units, but are the hierarchical level of taxonomic unit. They are termed as “operational taxonomic units (OTU’s)” [see (Sokal and Sneath 1963, p. 121) and (Sneath and Sokal 1973, p. 69)]. Here, OTUs or “the organizational level of a unit character” was used to define the primary taxonomic entities (taxa/species/individuals), which may be used as exemplars of taxonomic groups (Sneath and Sokal 1962). In the beginning, Sokal and Sneath used the term “operational taxonomic unit(OTU)” for the unit that is to be classified (Sokal 1963, p. 111), but deliberately used the term OTU’s instead of OTUs for representing the “operational taxonomic units” (Sokal 1963; Sneath and Sokal 1973). The term OTUs was used for representing “operational taxonomic units (OTUs)” in later time.” Such as the term “operational taxonomic units (OTUs)” was used in Sokal 1986’s review paper (Sokal 1986).
First it makes numerical taxonomy to get rid of relying on the validity of species defined by the conventional methods to define taxa. Thus, numerical taxonomy does not care how many species concepts and species definitions already have so far (Sokal and Sneath 1963, p. 121).
Second, it emphasizes that most taxa are phenetic, rather than to satisfy the genetic definition of species status (Sokal and Sneath 1963, p. 121). Thus, the concept of OTU is different from Mayr’s genetic definition of species (Mayr 1999) (first published in 1942).
Third, the term “taxon” (plural taxa) is retained for its proper function; that is, we can use the term taxon to indicate any sort of taxonomic group (Sneath and Sokal 1962), i.e., taxa are groups of OTUs or an abbreviation for taxonomic group of any nature or rank (Sneath and Sokal 1973). Thus, a taxon can be used to denote a variable for one or more characters, i.e., used as OTU’s higher taxa (e.g., genera, families, and orders) (Sokal and Sneath 1963, pp. 121–122).
7.4.2 Estimation of Taxonomic Resemblance
In Sokal and Sneath (1963), “resemblance,” “similarity,” and “affinity” are used interchangeably to imply a solely phenetic relationship. However, since affinity means kinship, hence by descent. Thus in Sneath and Sokal (1973), to avoid confusion the term “affinity” was not used; instead the terms “resemblance” and “similarity” are used interchangeably. Similarly the term relationship was used (see Sect. 7.5.1).
7.4.2.1 Data Matrix/OTU Table
Numerical taxonomy data matrix (or OTU table)
Characters | OTUs | |
|---|---|---|
(Attributes/descriptors/variables) | (Taxa/species/individuals/specimens/organisms/stands/analysis units/observations) | |
1 2 3 | A B C D E (Group A) | F G H I J (Group B) |
Ecological data matrix
Objects | Descriptors | |
|---|---|---|
(Sampling sites/sampling units/observations/locations/subjects/individuals/stands/quadrats/“taxa” such as woodland, prairie, or moorland in plant ecology) | (Species/attributes/variables) | |
1 2 3 | A B C D E (Group A) | F G H I J (Group B) |
Microbiome abundance data matrix
OTUs/ASVs | Samples | |
|---|---|---|
(Taxa/species/variables) | (Subjects) | |
1 2 3 | A B C D E (Group A) | F G H I J (Group B) |
Samples | OTUs/ASVs | |
|---|---|---|
(Subjects) | (Taxa/species/variables) | |
1 2 3 | A B C D E (Group A) | F G H I J (Group B) |
In numerical taxonomy data matrix (or OTU table) (Table 7.1), rows represent characters (attributes/descriptors/variables), while columns represent OTUs (taxa/analysis units/observations).
The setting of ecological data matrix is different from that in a numerical taxonomic matrix, in which rows represent objects (subjects/stands/quadrats), while columns represent descriptors (species). Although data matrices in ecology and numerical taxonomy have different structures (Legendre and Legendre 2012, p. 60), however, knowing the distinctions between attributes and individuals is very important because it is often confused between attributes and individuals in ecology. In ecology, attributes are species and individuals are objects (Table 7.2).
The structures of microbiome abundance data matrix (or OUT/ASV table) are relatively simple. It is a hybrid of numerical data matrix and numerical taxonomic matrix, in which rows represent OTUs (ASVs/sub-OTUs/variables), while columns represent samples (subjects) or reverse (Table 7.3). Here samples are analysis units; OTUs or species are variables. We do not need to distinct attributes and individuals. However, when we use microbiome software, we need to specify which data matrix structure is used. For example, the phyloseq package has been developed based on ecological software vegan package. When creating data object, it asks the user to specify whether taxa are_rows = TRUE or FALSE, which is confirming the structure of data matrix: whether rows represent taxa/OTUs/ASVs. In its initial version, the phyloseq package even used species to stand for taxa.
7.4.2.2 R-Technique and Q-Technique
Data matrices in Tables 7.1, 7.2 and 7.3 can be examined using two kinds of techniques (Cattell 1952): the R-technique and the Q-technique (originated by Stephenson under the name of inverted factor technique (Stephenson 1936, 1952, 1953)). In general, in an association matrix, a so-called Q matrix is formed to compare all pairs of individuals, while a so-called R matrix (dispersion or correlation matrix) is formed for comparisons between the variates. Numerical taxonomy starts logically with the choice of the t entities to be classified (OTUs) and n attributes or variables (generally characters in terms of biological work), which gives the n x t matrix. The R-technique examines the association of pairs of characters (rows) over all OTUs (columns), while the Q-technique, in contrast, examines the association of pairs of OTUs (columns) over all characters (rows). In numerical taxonomy, the R-technique refers to correlations among characters based on OTUs, considering the correlation of pairs of characters present in a population, while the Q-technique refers to the qualifications of relations between pairs of taxa, frequently species, based on a preferably large number of characters (Sokal 1963, pp. 125–126), considering the correlations of pairs of individuals present in a population in terms of their characters. In other words, Q-technique refers to the study of similarity between pairs of OTUs, and R-technique refers to the study of similarity between pairs of characters (Sneath and Sokal 1973, p. 256).
The adoption of using the R-technique and the Q-technique in numerical taxonomy has been criticized at the beginning after the publication of Sokal and Sneath’s 1963 book. The duality of the R or Q problem is one of some fundamental (Williams and Dale 1966) and statistical (Sneath 1967) problems in numerical taxonomy. For example, factor analysis compares characters but finally results in a grouping of OTUs. To overcome the confusion over the use of Q and R technique, Williams and Dale (1966) suggested that (1) Q and R should refer to the purpose of the analysis, i.e., refer to the matrix, and (2) A-space and I-space rather than R-space or Q-space should be used that is operated upon when the relationships are represented in a hyperspace.
The organisms can be represented as points in a multivariate character space (A-space, or attribute space), and conversely the characters can be represented in an I-space (individual space) (Sneath 1967). A-space and I-space refer to the model: the former is used for a model in attribute-space and the latter is used for a model in the individual-space. A matrix of inter-individual distances implies relationships between points in an A-space, while an inter-individual correlation matrix implies angles in an I-space. They both are Q-technique. An attribute-correlation matrix is R-technique which concerns with angles, while an individual-distance matrix is Q-technique which concerns with point.
Sneath (1967; Sneath and Sokal 1973) accepted the two suggestions of Williams and Dale (1966). In numerical taxonomy, R-technique leads to a classification of characters, Q-technique to one of OTUs. But they emphasized formally both R-technique and Q-technique have the same main mathematical steps because by transposing the data matrix in an R study, the characters (rows) become the individuals comparable to the former OTUs, and the actual OTUs or taxa (columns) become the characteristics (attributes) over which association is computed (Sneath and Sokal 1973, p. 116). In either space the character values are the coordinates of the points in the hyperspace. Based on the distinction between A-space and I-space in Williams and Dale (1966), in numerical taxonomy, A-space (attribute space) has formally n dimensions (one for each attribute or character) with t points that represent the OTUs. I-space (individual space) has formally t dimensions (one for each OTU) with n points representing the attributes or characters (Sneath and Sokal 1973) although less than n (in A-space) or t (in I-space) dimensions respectively could be after attempting the dimensionality reduction (p. 116).
Cattell (1966) restricted Q- and R-techniques to factor analysis, and applied Q-technique as a customary practice to numerical taxonomy in terms of cluster analysis of OTUs. Both Q- and R-techniques are employed in cluster as well as factor analysis although numerical taxonomy mainly emphasized the Q-technique. By using the notations of A-space and I-space, most numerical taxonomic methods are operated by Q-technique on an A-space, but some cluster and factor analyses are Q-techniques operating on an I-space.
Overall, Sneath and Sokal emphasized that Q- and R-techniques have been adapted in the general sense in numerical taxonomy. Conveniently a Q-study is used for the quantification of relations between organisms according to their characters aiming to produce classifications of organisms; while an R-study refers to classifying characters into groups according to the organisms that possess them, in which the characters become in effect the OTUs, and the procedures are similar to Q-study, but operate on the transpose of the n x t matrix [See (Sneath 1967; Sneath and Sokal 1973, pp. 115–116)].
Basically, the uses of Q- and R-techniques (or analyses) in numerical taxonomy have been adopted in numerical ecology (Legendre and Legendre 2012, pp. 266–267), whereas Q measures (Q mode study) are used for the association coefficients (the relationships) among objects given the set of descriptors and R measures (R mode analysis) are used for the association coefficients (the relationships) among descriptors (variables) for the set of objects in the study.
In summary, Q analyses refer to the studies that start with computing an association matrix among objects, whereas R analyses refer to the studies that start with computing an association matrix among descriptors (variables). Numerical taxonomy defines a character as a variable (feature) and OTU as an analysis unit, which leads to the different understanding of R-technique and Q-technique in some other taxonomists and ecology. Later microbiome study adopted the methods of numerical taxonomy, but not the definition of character, where samples are observations and OTUs are variables (features/characteristics). The way data matrix is used in microbiome study is consistent with ecology. In microbiome study, Q- and R-techniques (or analyses) are not explicitly differentiated but in some software that are adopted from ecology the terms and according data matrices are often used. In terms of R-technique for relationship among variables and Q-technique for relationship among individuals, the microbiome study mainly uses R-technique to find the relationship among variables (OTUs/ASVs/taxa) over the samples.
7.4.2.3 Similarity Coefficients
In numerical taxonomy, the term “similarity coefficient” is most commonly used, although in the strict sense the term “coefficient of resemblance” would be more appropriate. The term “coefficients of similarity” or “coefficients of dissimilarity” is also used. Numerical taxonomy roughly groups four types of coefficients—distance coefficients, association coefficients, correlation coefficients, and probabilistic similarity coefficients. They collectively refer to as coefficients of resemblance or similarity and are used to compute resemblances between taxa.
The space that measures similarity does not need to be Euclidean in the strict sense, but it should be metric, in other words, determined by a metric function (Williams and Dale 1966; Johnson 1970).
Based on the requirements for similarity coefficients, Sneath and Sokal (1973) described the four axioms over the entire set of OTUs that the measures of dissimilarity or functions (converting similarity coefficients into measures of dissimilarity) should be satisfied (p. 120). This is very important when we apply these similarity coefficients or develop the new measures of similarity. However, in microbiome literature, this topic has been rarely mentioned. Thus, here we briefly describe these four axioms before we formally introduce similarity coefficients.
Axiom 1: φ(a, b) ≥ 0, and φ(a, a) = φ(b, b) = 0. It states that identical OTUs are indistinguishable while nonidentical ones may or may not be distinguishable by the dissimilarity function.
Axiom 2: φ(a, b) = φ(b, a). This is the symmetry axiom. It states that the value of function φ from a to b is the same as from b to a.
Axiom 3: φ(a, c) ≤ φ(a, b) + φ(b, c). This is the triangle inequality axiom. It states that the function between a and c cannot be greater than the sum of the functions between a and b and b and c. In other words, the sum of the lengths of two lines is greater than the length of third line.
Axiom 4: If a ≠ b, then φ(a, b) > 0. This axiom states that if a and b differ in terms of their character states, then the function between them must be greater than zero.
All these four axioms are true for the Euclidean distances (which additionally obeys Pythagoras’ theorem: a2 + b2 = c2, i.e., the lengths of the square whose side is the hypotenuse (the side opposite the right angle) is equal to the sum of the lengths of the squares on the triangle’s other two sides), and thus they are metrics. However, not all measures of dissimilarity are metrics in numerical taxonomy. For example, there exist the cases that the difference in character states between two OTUs a and b is known; however, the function between a and b may still be zero. Thus, the axiom 4 is not fulfilled for all OTUs in the taxon (which are called as pseudometric or semimetric systems) (Sneath and Sokal 1973, p. 121). For ease of understanding of the relationships, numerical taxonomy mainly focuses on the coefficients whose properties lead to metric spaces.
Axiom 3*: φ(a, c) ≤ max [φ(a, b), φ(b, c)].
Here the pair-function is called an ultrametric pair-function for any pair of OTUs. A phenogram is one example of this function (Sneath and Sokal 1973, p. 121).
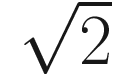 in two-dimensional model, would be
in two-dimensional model, would be 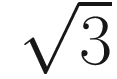 in three-dimensional model, and
in three-dimensional model, and 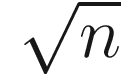 in an n-space, a hyperspace of n dimensions (Sokal 1963, pp. 143–144). Table 7.4 summarizes some measures of taxonomic distance compared by Sokal and Sneath.
in an n-space, a hyperspace of n dimensions (Sokal 1963, pp. 143–144). Table 7.4 summarizes some measures of taxonomic distance compared by Sokal and Sneath.Selected measures of taxonomic distance in numerical taxonomy
Definition | Reference and comments |
|---|---|
The mean character difference of (Cain and Harrison 1958):
| It is defined as the absolute (positive) values of the differences between the OTUs for each character It is a special case of Manhattan or city-block metric It has the advantages of its simplicity and it is a metric It also has some disadvantages, such as always underestimating the Euclidean distance between the taxa in space, lacking of the desirable attributes of the taxonomic distance or its square (Sneath and Sokal 1973) |
The taxonomic distance of (Sokal 1961):
And the average distance is defined as:
| The taxonomic distance between two OTUs j and k is defined in (Sokal 1961; Rohlf 1965; Sokal 1963) This is a generalized version of the Euclidean distance between two points in an n-dimensional space Like to M. C. D., it is a special case of Manhattan or city-block metric It was first used by Heincke as early as 1898 (Sokal and Sneath 1963)
The expected value of d very closely approaches |
The coefficient of diverge of (Clark 1952):
| The ratio varies between zero and unity Originally each OTU represents a number of specimens, while single values are used in numerical taxonomy (Sokal 1963) |
The coefficient of racial likeness (Sokal and Sneath 1963):
| This formula was developed based on Karl Pearson (1926)’s coefficient of racial likeness (Pearson 1926), in which the rows of the Q-type matrix are standardized The OTUs are represented by only single values (with a variance of one) rather than means of the ith character for entity j (Sokal 1963) The strengths of this coefficient (Sneath and Sokal 1973, p. 185) are (1) taking the variance of the estimates into consideration and (2) permitting estimates of the degree of misclassification of individuals in two populations |
The generalized distance developed by Mahalanobis (1936) and Rao (1948):
Where W−1 is the inverse of the pooled variance-covariance (dispersion) matrix within samples with dimension n × n δJK is a vector of differences between means of samples J and K for all characters | The generalized distance is equivalent to distances between mean discriminant values in a generalized discriminant function, and therefore measuring the distance as a function of the overlap between pairs of populations and transforming the original distances so as to maximize the power of discrimination between individual specimens in the constructed groups (i.e., the OTUs) (Sneath and Sokal 1973, p. 128) It is computed by first obtaining the maximal variance between pairs of groups relative to the pooled variance within groups and then maximizing the difference between pairs of means of characters (Sneath and Sokal 1973, p. 127) The generalized distance has the same strengths as the coefficient of racial likeness (Sneath and Sokal 1973, p. 185): (1) accounting for the variance of the estimates into consideration and (2) permitting estimates of the degree of misclassification of individuals in two populations It is particularly useful in numerical taxonomy (Sneath and Sokal 1973, p. 127) However, it also has some weaknesses (Sneath and Sokal 1973, p. 128): (1) it may distort the original distances considerably; (2) small eigenvalues may make a large haphazard contribution to the distance when a large number of characters is employed; (3) its value is heavily dependent on assumptions of multivariate normal distributions within the OTUs, which will usually not hold above the population or species level |
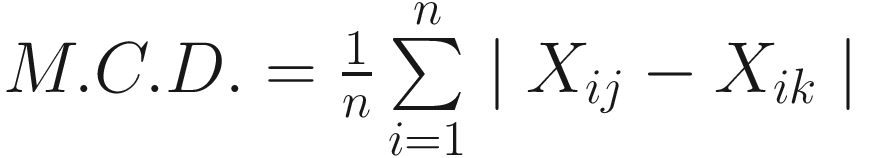
![$$ {\Delta}_{jk}={\left[\sum \limits_{i=1}^n{\left({X}_{ij}-{X}_{ik}\right)}^2\right]}^{1/2}, $$](images/486056_1_En_7_Chapter_TeX_IEq5.png)
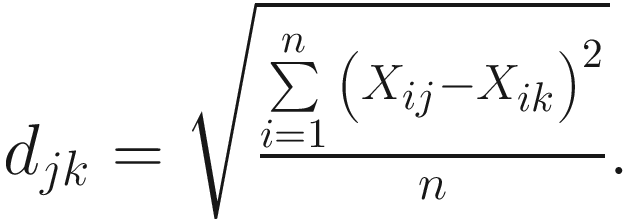
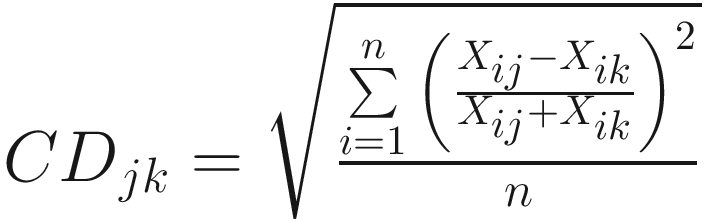
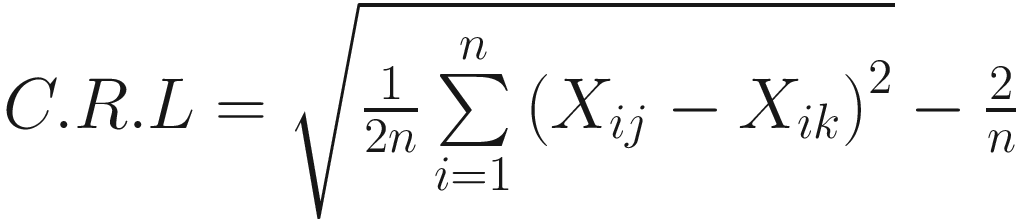
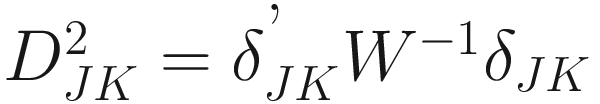
They are used for categorical data (two-state characters or multi-state characters) to measure the agreement of the states in the two data columns representing the OTUs in numerical taxonomy.
The association coefficients are also called similarity coefficients, relationship or matching coefficients. In a Q-type 2 × 2 data matrix or table, a parametric resemblance value between the two OTUs is expressed as a percentage of matching characters. It measures a sample proportion (percentage) of matches from an infinitely large population of character matches which could be attempted (Sokal and Sneath 1963). Three kinds of association coefficients have been proposed for use in numerical taxonomy.
The 2 × 2 table for computing association coefficients
OTU(Taxon) j | ||||
1 | 0 | |||
OTU(Taxon) k | 1 | a | b | a + b |
0 | c | d | c + d | |
a + c | b + d | n = a + b + c + d | ||
Selected coefficients of association for two-state characters in numerical taxonomy
Definition | Reference and comments |
|---|---|
The coefficient of (Jaccard 1908):
| As used by (Sneath 1957a, b; Jaccard 1908) Excludes negative matches in its calculation of coefficient This is the simplest of the coefficients, which has been widely used in R-type and Q-type ecology studies It is appropriate if without considering inclusion of the negative matches in its calculation because it is simplest coefficient (Sokal 1963, p. 139) The values of the coefficient of Jaccard range between 0 and 1 It has three disadvantages (Sneath and Sokal 1973): (1) the complements of SJ coefficient are nonmetric (pseudometric), (2) do not necessarily obey the triangle inequality, and (3) do not consider matches in negative character states (actually the coefficient of Jaccard is appropriate when excluding negative matches is considered as appropriate) |
The simple matching coefficient (Sokal and Michener 1958):
| First suggested by Sokal and Michener (1958) Equals to the coefficient of Jaccard but includes the negative matches Restricts to dichotomous characters only If considering all matches, Sokal and Sneath preferred the simple matching coefficient over that of Rogers and Tanimoto because it is simpler and easier to interpret (Sokal 1963, p. 139) |
The coefficient of (Rogers and Tanimoto 1960):
| This similarity ratio was originally developed to include characters with more than two states and account for missing information Sokal and Sneath limited this coefficient to the case of two states per character and with complete information (Sokal 1963, p. 134) It includes the negative matches |
The coefficient of (Hamann 1961):
| Balances the number of matched and unmatched pairs in the numerator The values rang from −1 to +1 This coefficient has an undesirable property and independence probably does not have a clear meaning in 2 × 2 table in numerical taxonomy (Sokal 1963, p. 134) The concept of balancing matches against mismatches does not appear of special utility in the estimation of similarity (Sneath and Sokal 1973, p. 133) |
The coefficient of (Yule and Kendall 1950):
| Balances the number of matched and unmatched pairs in the numerator The values rang from −1 to +1 It has rarely been employed in numerical taxonomy (Sneath and Sokal 1973, p. 133) |
The phi coefficient (Yule and Kendall 1950):
| Balances the number of matched and unmatched pairs in the numerator The values rang from −1 to +1 It is important because of its relation to χ2 (χ2 = ϕ2n), which permits a test of significance (Sokal 1963, p. 135) However, the test of significance is unmeaningful due to the problem of heterogeneity of column vectors (Sokal and Sneath 1963, p. 135) |
The general similarity coefficient of (Gower 1971):
This is one version of Gower’s coefficient for two individuals j and k, which is obtained by assigning a score 0 ≤ Sijk ≤ 1 and a weight wijk for character i The weight wijk is set to 1 for a valid comparison for character i and to 0 when the value of the state for character i is unknown for one or both OTUs | It is applicable to all three types of characters: two-state, multistate (ordered and qualitative), and quantitative (Sneath and Sokal 1973, pp. 135–136) In a data matrix consisting of two-state characters only, Gower’s general coefficient therefore becomes the coefficient of Jaccard (SJ). Typically Sijk is 1 for matches and 0 for mismatches and sets wijk= 0 when two OTUs match for the negative state of a two-state character For multistate characters (ordered or qualitative) Sijk is 1 for matches between states for that character and is 0 for a mismatch. Due to without consideration of the number of states in the multistate character, the coefficient resembles a simple matching coefficient applied to a data matrix involving multistate characters For quantitative characters Gower sets Sijk = 1 − (|Xij − Xik|/Ri), where Xij and Xik are scores of OTU j and k for character i, and Ri is the range of character i in the sample or over the entire known population. It results in Sijk= 1 when character states are identical and Sijk= 0 when the two character states in OTUs j and k span the extremes of the range of the character The general similarity coefficient of Gower has the advantages: (1) these coefficient matrices are positive semi-definite which make it possible to represent the t OTUs as a set of points in Euclidean space (Sneath and Sokal 1973, p. 136); (2) can obtain a convenient representation by taking the distance between the jth and kth OTUs proportional to However, this similarity coefficient also has the disadvantage: the positive semidefinite property of the similarity matrix could be lost due to NCs (no comparisons) (this is also true of R-type or character- correlation matrices involving substantial numbers of NCs) (Sneath and Sokal 1973, p. 136) |
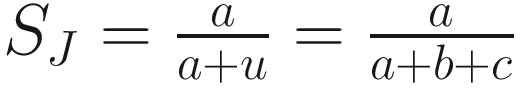
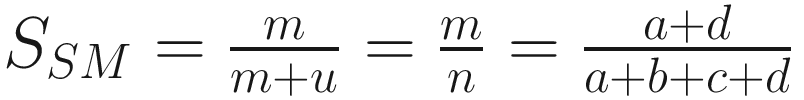
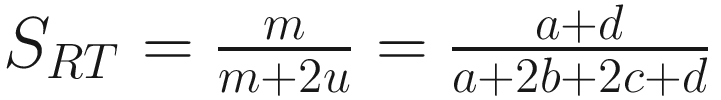
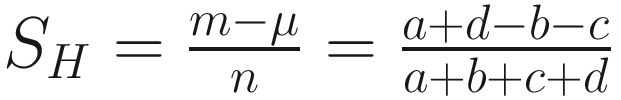
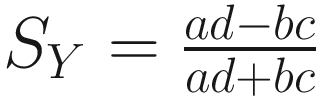
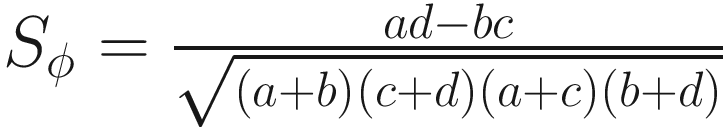
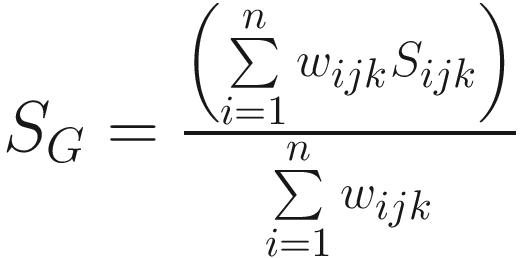

In summary, generally the association coefficients include both positive and negative matches in their calculation of coefficients. Both similarity and dissimilarity coefficients are used for measuring resemblance. For binary variables, Jaccard (similarity) coefficient was used in Sneath (1957b) and single-matching coefficient was used in Sokal and Michener (1958). Sneath and Sokal (1973) advocated negative matches and hence favored the modified Gower’s coefficient which allows negative matches in two-state characters. They thought that the modified Gower’s coefficient appeared to be a very attractive index for expressing phenetic similarity between two OTUs based on mixed types of characters (p. 136).
Correlation coefficients are special cases of angular coefficients. In numerical taxonomy, correlation coefficients are used to measure proportionality and independence between pairs of OTU vectors, in which both the product-moment correlation coefficient for continuous variables and other correlation coefficients for ranks or two-state characters have been employed. The product-moment correlation coefficient is most often used in early numerical taxonomy; other correlation coefficients such as the rank correlation have also been occasionally used (Daget and Hureau 1968; Sokal 1985).
For continuous variables, the Pearson product-moment correlation coefficient was most often used in both psychology and ecology Q-type studies previously (Stephenson 1936). At the end of 1950s, it was first proposed to use in numerical taxonomy by (Michener and Sokal 1957; Sokal and Michener 1958), and has been used since then and become the most frequently employed similarity coefficients in numerical taxonomy for calculating between pairs of OTUs (Sneath and Sokal 1973, p. 137). The coefficient, computed between taxa j and k, is given as:
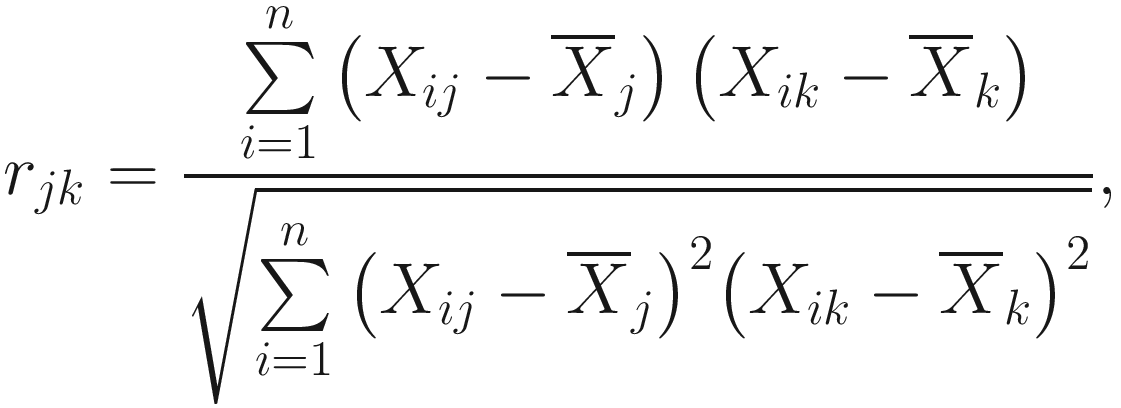
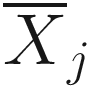 is the mean of all state values for OTU j, and n is the number of character sampled. The values of correlation coefficients range from −1 to +1.
is the mean of all state values for OTU j, and n is the number of character sampled. The values of correlation coefficients range from −1 to +1.- (1)
Theoretically it is possible that high negative correlations between OTUs exist, but practically they are unlikely in actual data (Sneath and Sokal 1973, p. 138). The argument for this statement is that Sneath and Sokal thought that a study has a few OTUs, then “pairs of OTU’s antithetical for a sizeable number of characters are improbable” (Sneath and Sokal 1973, p. 138). It actually stated that the components of OTUs are not compositional.
- (2)
Pearson correlation analysis is based on the bivariate normal frequency distribution. However, numerical taxonomic data are not normally distributed. Thus, it cannot be directly applied into numerical taxonomic data (Sneath and Sokal 1973, p. 138). Three reasons that make numerical taxonomic data unlikely to be normally distributed: (i) the heterogeneity of the column vectors; (ii) dependence of characters (rows of the data matrix); and (iii) different characters being measured in widely varying scales. Therefore it is invalid to conduct statistical hypothesis testing of correlation coefficients in numerical taxonomy based on the usual assumptions.
- (3)
For this reason, numerical taxonomists generally advocated standardization of the rows of the data matrix (i.e., standardization of characters over all OTUs) to make the mean of the column vectors frequently approaches 0 and to allow representing inter-OTU relations by using a relatively simple geometrical model (Rohlf and Sokal 1965; Sneath and Sokal 1973, p. 138). The goal of standardization of the characters is to make the vectors carrying the OTUs to be unit length and the column means to be zero (i.e., approximately normalize the column vectors of the OTUs), in such that correlations between OTUs are functions of the angle between the lines connecting them to the origin. Thus, the cosine of the angle between any two vectors is an exact measure of the correlation coefficient (Sneath and Sokal 1973, pp. 138–139).
(1) They are generally nonmetric functions although the semichord distances can be transformed to yield a Euclidean metric. When correlation coefficients are converted to their complementary form (i.e., distances), generally the axiom of triangle inequality is violated (Sneath and Sokal 1973, p. 138). Perfect correlation could also occur between nonidenticals, such as in two column vectors when one is the other multiplied by a scalar. (2) The correlation coefficient in numerical taxonomy has an undesirable property because in the cases with few characters, the values of the correlation coefficient will be affected by the direction of the coding of the character states (Eades 1965). Thus, it requires that all characters have the same directional and dimensional properties (Minkoff 1965).
However, overall Sneath and Sokal (1973, p. 140) believe that correlation coefficients are usually the most suitable measure in numerical taxonomy to evaluate OTUs. They do not think above undesirable properties would not manifest themselves in large data matrices involving many OTUs with the large number of characters. Additionally, when the interpretation of taxonomic structure is based on phenograms, it has been observed that the correlation coefficients are the most appropriate measure.
In summary, due to heterogeneity of OTUs, the basic assumptions of the bivariate normal frequency distribution cannot be met, so in psychology and numerical taxonomy literature, the character scores are often standardized or a percentage scale or a ratio of the variable are used before applying this coefficient (Sokal and Sneath 1963, p. 142).
Probabilistic similarity coefficients were developed in 1960s; Sneath and Sokal have reviewed these similarity coefficients in their 1973 book (Sneath and Sokal 1973).
First we should point out that in numerical taxonomy, the characters are the variables; OTUs are observations. Thus, here, the distribution refers to the distribution of the frequencies of the character states over the set of OTUs. While as Sneath and Sokal (1973) pointed out, probabilistic similarity indices are more relevant to ecological classification, where the abundance of a species (species are the equivalent of characters in numerical taxonomy, and stands correspond to OUTs) in a sampling unit is a stochastic function (p. 141).
Probabilistic similarity coefficients were developed based on the assumption that it is less probable to agree among rare character states than to agree for frequent character states and therefore rarer character states should be given more heavy or enhanced weight (Sneath and Sokal 1973, p. 140). The technique of probabilistic similarity had been developed to avoid using similarity coefficients and instead to go straight to use the data matrix for a classification, in which the establishing partitions are guided by only a criterion of goodness of the classification (Sneath and Sokal 1973, p. 120).
is a similarity index to measure the complement of the probability that two OTUs would have the observed (or a greater) degree of similarity if attribute values were assorted at random. It was shown by Sneath and Sokal (1973) that this index resulted in very good general agreement with the analyses using simple Jaccard similarity indices.
One important kind of similarity coefficients is the information statistics. It has the advantage of additive property. Thus information statistics permits the partitioning of indices for larger groups into information statistic for subgroups (Sneath and Sokal 1973).
In ecology, information statistics is used to measure the frequencies or probabilities of the occurrence of the various species over the collection of stands. Sneath and Sokal introduced one probabilistic similarity coefficient based on information-theoretical concepts into numerical taxonomy to measure the homogeneity of the system by partitioning or subpartitioning sets of OTUs (Sneath and Sokal 1973, p. 120).
was developed based on information statistics. Technically “information” is a measure of disorder, variance, or confusion, or “surprisal” as Orloci (1969) put out, rather than a measure of the amount of knowledge about a group. Given a probability distribution for the occurrence of various character states over the t available OTUs (Goodall 1966b), the measure of disorder for character i based on a measure of information or entropy is defined as follows.
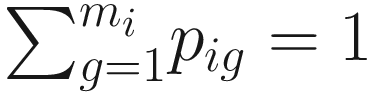 . When the n characters are not correlated, the total information of the group can be obtained by summing the separate values of
. When the n characters are not correlated, the total information of the group can be obtained by summing the separate values of 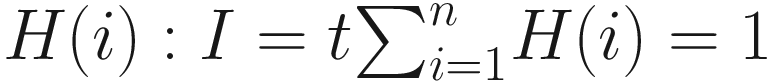 .
.The constructed information or entropy can also be applied to ecological data. In an ecological setting characters (rows) are species and OTUs (columns) are stands or quadrats, and hence for data matrices in ecology, this formula is to measure the frequencies of any one species that are recorded over a series of stands (Sneath and Sokal 1973, p. 142). To conduct hypothesis testing of “states” mi, these frequencies must be grouped into classes to provide mi “states” (e.g., “absent,” “rare,” “common,” and “abundant”) of a species (or character) i.
An approximately x2 distribution with 2n degrees of freedom can be described by this formula in terms of frequencies for taxon J containing tJ OTUs. Where the quantity I measures the total information (entropy, surprisal) in the entire sample with the lower value of I indicating the more homogeneous a taxon will be. Thus, one goal of clustering OTUs using information statistics is to obtain taxa with low values of I (Sneath and Sokal 1973, p. 142).
Compared to most resemblance coefficients that only apply to pairs of single OTUs or pairs of taxa, another advantage of the information statistics is it’s applicable to sets of OTUs. Sneath and Sokal (1973, pp. 143–145) introduced an index based on two concepts: joint information and mutual information discussed by (Estabrook 1967; Orloci 1969). Given the correlation of characters, joint information I(h, i) is the union of the information content of two characters over a set of OTUs; mutual information I(h; i) is their intersection. The relation between these quantities is given below:
The mutual information I(h; i) is the ratio of the sums of the information content exclusive to characters h and i divided by the total information possessed jointly by h and i.
Overall, information statistics have important properties (Sneath and Sokal 1973, p. 120), such as are usually additive, are distributed as chi-square, and are probabilistic, and therefore a statistical hypothesis testing can be conducted through a standard statistical means. Additionally, the special cases of these coefficients can be represented as distance coefficients.
The denoising methods that are used to classify ASVs or sub-OTUs in current microbiome research actually take the same approaches as in probabilistic similarity coefficients (see Chap. 8 for details).
The choice of which resemblance coefficient not only is determined by the nature of the original data matrix, such as association coefficients for binary and categorical data matrices and distance and correlation for continuous variables, but also is based on the type of resemblance. Sokal (1985) reviewed that classifications have been based on correlation coefficients than based on similarity using distance-based measures. For binary data, correlations are not especially helpful and standardization does not help either.
In summary, regarding these three coefficients of association, distance measures, and correlation, Sokal and Sneath (1963, p. 167) thought that association and distance are easier to interpret conceptually and relatively simpler than correlation. Among these three coefficients, they recommended using the simple matching coefficient and Jaccard coefficient in case of excluding negative matches, the coefficient of taxonomic distance djk based on standardized characters, and Pearson’s product-moment rjk, respectively. In their 1973 book Sneath and Sokal favor more strongly than in their 1963 book on binary similarity coefficient because of the following: (1) its simplicity and (2) possible relationship to information theory; (3) the similarity between fundamental units of variation can be estimated; and (4) related to natural measures of similarity or distance between fundamental genetic units (amino acid or nucleotide sequences) (Sneath and Sokal 1973, p. 147).
7.4.3 Commonly Clustering-Based OTU Methods
Taxa (taxonomic structure/taxonomic system) are constructed through resembling similarity (the resemblance matrix) at the same or different rank levels hierarchically by using a measure of resemblance, i.e., grouping together OTUs to form taxa. The taxonomic system that is to be constructed should be “natural” in an empirical sense, and thus should be highly predicable and be formed with a nested hierarchy.
7.4.3.1 Similarity/Resemblance Matrix
Resemblance matrix of OTUs
OUTs | OUTs | |||||
1 | 2 | 3 | … | t | ||
1 | S11 | S12 | S13 | … | S1t | |
2 | S21 | S22 | S23 | … | S2t | |
3 | S31 | S32 | S33 | … | S3t | |
⋮ | ⋮ | ⋮ | ⋮ | ⋮ | ||
t | St1 | St2 | St3 | … | Stt | |
Lower left triangle resemblance matrix of OTUs
OUTs | OUTs | |||||
1 | 2 | 3 | … | t | ||
1 | ||||||
2 | S21 | |||||
3 | S31 | S32 | ||||
⋮ | ||||||
t | St1 | St2 | St3 | … | ||
The main techniques used for constructing taxonomic structure are clustering techniques, which are applicable to all three types of similarity coefficients (distance, association, and correlation coefficients). The more complicated method factor analysis can be used to construct taxonomic structure.
Four sets of methods have been developed in numerical taxonomy: (1) clustering, (2) factor analysis, (3) ordination, and (4) discriminant analysis. We can collectively call them as “numerical taxonomic methods” or “OTU methods” since they are used to classify or identify the OTUs. The first two sets of methods were mainly proposed or developed in Sokal and Sneath’s early work, such as in their 1963 book,whereas the last two sets of methods were mainly adopted into numerical taxonomy in their later works (e.g., 1973 book). The first three sets of methods are used for classification, and the discriminant analysis is used for both classification and identification. We describe them separately below.
One goal of clustering in numerical taxonomy is to recognize the patterns of distribution of OTUs and groups of OTUs (taxa) in a space (commonly a hyperdimensional, phenetic A-space) (Sneath and Sokal 1973, p. 192).
7.4.3.2 Definitions of Cluster in Numerical Taxonomy
When the patterns are descripted, they are typically conducted at least within a three-space (mathematically a higher-dimensional space): multidimensional Euclidean, non-Euclidean, and nonmetric spaces. Generally, regular and random distributions are the two observed dispersion patterns that can be compared. The clusters in numerical taxonomy are defined as the sets of OTUs in phenetic hyperspace that exhibit neither random nor regular distribution patterns and that meet one or more of various criteria.
Cluster definitions in numerical taxonomy
Definition and description |
|---|
Cluster center The OTUs are within the center of average organism or centroid For the hypothetical OTU (the hypothetical modal organism), it should be at the corner that is nearest to the centroid For an actual OTU, it is the centrotype. That is the OTU with the highest mean resemblance to all other OTUs of the cluster or the OTU nearest to the centroid for Euclidean distance models, or the OTU having the highest loading on a factor representing the cluster in a Q analysis |
Density A cluster should be denser than other areas of the space under consideration It can be expressed as number of OTUs per unit hypervolume or as the density/volume ratio of a convex hyperdimensional set |
Variance A cluster for a satisfactory partition should be that variance within the subsets is minimized relative to variance among the subsets in analysis of variance (univariate and multivariate) |
Dimension A dimension of a cluster can be measured in various ways In discriminatory methods the cluster is defined in terms of a radius of the cluster, i.e., a specified majority of OTUs lie within this radius In ordination methods, the ellipticity of a cluster can be estimated from the standard deviations of the OTUs on the several factor axes (the square roots of the eigenvalues) |
Number of members in a cluster Some clustering methods either Allocate weights according to the number of members in a cluster, or Require that a group shall contain a given minimum or maximum number of OTUs (the cluster size) to recognize a cluster |
Connectivity To be members of a cluster, individuals have to be related (i.e., connected) to a certain minimal degree, such as within some kind of closeness by distance in the hyperspace It requires that each OTU is connected at the minimal acceptable level to every other OTU either directly or via other OTUs |
Straggliness It implies much greater extension in a few of the dimensions than in the others Ellipticity can be revealed by examining factor analysis plots, or calculating the standard deviations The highly straggly clusters can also be shown by a histogram of distances of OTUs from the centroid to see whether a sharp mode is well away from the origin, or is flattened, falling off from the origin |
Gaps and moats The gap or moat by which the distance from each member of a cluster to the closest one in another subset should be sufficiently large to separate or isolate the subsets A moat can be the absolute distance between the closest members of different subsets, or the average of such distances for all members of both subsets; or may be some ratio of the distance between the centroids of clusters and the radial vectors of the cluster envelopes to their centroids The gap between clusters is a criterion of measuring a new type of cluster, for which the greatest distance between any pair of OTUs within the cluster is less than the smallest gap from a cluster member to any OTU outside the cluster |
7.4.3.3 Commonly Clustering Methods in Defining Groups of OTUs
Numerical classification can be performed by arranging of organisms in one of three ways (Sneath and Sokal 1973, p. 200): (1) hierarchical clustering systems of partitions (nested, mutually exclusive systems), (2) nonhierarchic clustering systems of partitions, and (3) ordination-the positioning of OTUs in relation to multidimensional character axes in an A space. In this section, we introduce clustering including both hierarchical clustering and non-hierarchic clustering. In the next section, we will briefly review ordination techniques in numerical taxonomy, and the more detailed introduction will be presented in Chap. 10 when microbiome study is covered.
In numerical taxonomy, cluster analysis is referred to as a large class of numerical techniques for defining groups of related OTUs based on high similarity coefficients. Clustering methods typically represent the similarity matrix among taxonomic units by dendrograms (a treelike arrangement). The grouped OTUs by clustering may form a coarser partition from a finer one, or may separate an entire set of OTUs into increasingly finer partitions.
Clustering can be described from eight aspects: (1) agglomerative versus divisive; (2) hierarchic versus non-hierarchic; (3) non-overlapping versus overlapping; (4) sequential versus simultaneous; (5) local versus global criteria; (6) direct versus iterative solutions; (7) weighted versus unweighted; and (8) non-adaptive versus adaptive. Among these eight methods, the sequential, agglomerative, hierarchic, non-overlapping (the acronym SAHN) clustering methods are the most frequently employed strategies for finding clusters (Sneath and Sokal 1973, p. 214).
The grouping of OTUs in numerical taxonomy has been most often employed by sequential clustering and by agglomerative techniques and rarely by divisive techniques. Agglomerative techniques start with a set of t separate entities, group them into successively fewer than t sets, arriving eventually at a single set containing all t OTUs. In contrast, divisive techniques start with all t OTUs in one set, subdividing it into one or more subsets, until each of the subsets is subdivided. Numerical taxonomy defines hierarchies in terms of nestedness as well as mutual exclusiveness. Thus, hierarchic clustering is to classify organisms into nested, mutually exclusive taxa. The hierarchic classifications are referred to as stratified. In a non-overlapping method, taxa at any one rank are mutually exclusive; that is, if the OTUs have been contained within one taxon, they cannot also be members of a second taxon of the same rank. For the details, the interested readers are referred to Sneath and Sokal’s 1973 book (Sneath and Sokal 1973, pp. 202–214).
Here, we summarize the commonly SAHN clustering methods used for defining groups of OTUs. Among them, four mainly described cluster methods are (1) single linkage clustering, (2) complete linkage clustering, (3) average linkage clustering, and (4) central or nodal clustering.
7.4.3.3.1 Single Linkage Clustering
Single linkage clustering (Sneath 1957a) establishes connections between OTUs and clusters and between two clusters by single links between pairs of OTUs (Sneath and Sokal 1973, p. 218). First, it finds and clusters together those mutually most similar pairs (least dissimilar pairs) with the highest possible similarity coefficient. Then, it successively lowers the level of admission by steps of equal magnitude and examines distances from new candidates for fusion with the established clusters. Thus, in single linkage clustering, a candidate OTU for an extant cluster has similarity to that cluster equal to its similarity to the closest member within the cluster (Sneath and Sokal 1973, p. 218).
Single linkage clustering is also called clustering by single linkage (Sneath’s method) (Sokal and Sneath 1963) or the nearest neighbor clustering (Lance and Williams 1966) or minimum method (Johnson 1967). It was introduced to taxonomy by Florek et al. (1951a, b) and Sneath (1957a). Single linkage clustering has the disadvantages: (1) sensitive to noise in the data (Milligan 1996); (2) may form complicated serpentine clusters, which is not very desirable in biological taxonomy (Sokal and Sneath 1963, pp. 192–193); and (3) leads to a peculiar feature: chaining (elongate growth), which especially occurs when a number of equidistant points or near equidistant points are frequently found in taxonomy and ecology. Such chaining causes phenograms not to be very informative because the phenograms do not well show the information on intermediate or connecting OTUs (Sneath and Sokal 1973, p. 223).
7.4.3.3.2 Complete Linkage Clustering
Complete linkage clustering (Sørensen 1948) joints two clusters by their similarity that exists between the farthest pair of members, one in each cluster (Sneath and Sokal 1973, p. 222). It requires a given OTU joining a cluster at a certain similarity coefficient must have relation at that level or above with every member of the cluster. Thus, in complete linkage clustering, a candidate OTU for admission to an extant cluster has similarity to that cluster equal to its similarity to the farthest member within the cluster (Sneath and Sokal 1973, p. 222).
- (1)
Generally leads to tight, hypersphencal, discrete clusters that join others only with difficulty and at relative low overall similarity values (Sneath and Sokal 1973, p. 222).
- (2)
Likely forms very compact and well-defined clusters, which look like clouds of OTUs in a hyperspace and like hyperspheroids in that space (Sokal 1963, p. 192) or small tight compact clusters (Sneath and Sokal 1973, p. 223).
- (3)
Different initial levels of similarity coefficients could result in different number of clusters (Sokal 1963).
7.4.3.3.3 Average Linkage Clustering
Average linkage clustering is also called clustering by average linkage (the group method of Sokal and Michener) (Sokal 1963), and earlier it was called group methods in numerical taxonomy (Sneath and Sokal 1973). Average linkage clustering techniques were developed to avoid the extremes resulted in either single linkage or complete linkage clustering. This clustering can be based on all three types of similarity coefficient matrices although the correlation coefficient matrices are originally suggested. Sometimes Spearman’s sums of variables method (Spearman 1913) was used for calculation of the correlation coefficients with average linkage clustering in numerical taxonomy. The average cluster method is recommended for use because single and complete linkage clustering methods have their disadvantages (Sokal and Sneath 1963, pp. 192–193).
7.4.3.3.4 Central or Nodal Clustering
Central or nodal clustering (Rogers and Tanimoto 1960) starts with an association coefficient (similarity coefficient). It calculates the value Ri based on a matrix of similarity coefficients to represent the number of nonzero similarity coefficients that a given OTU i has with other OUTs. Ri value is considered as the similarity coefficients greater than the expected value (or a given criterion) to give useful clusters (Sokal and Sneath 1963). In practice, the Ri values, which are indices of typicality, are considered only similarity coefficients larger than 0.65 (Silvestri et al. 1962). The larger the Ri value, the more “typical” a given OTU is to group under the study.
Central or nodal clustering is also called the method of Rogers and Tanimoto (Sokal and Sneath 1963). It has the advantage to obtain a measure of the homogeneity of clusters because it computes a coefficient of inhomogeneity, which is desirable in biology (Sokal 1963, p. 193). However, it also has the disadvantages: (1) defines only a series of primary nodes without connecting them into a dendrogram, and (2) is much indeterminant to obtain the clustering solutions (Sokal and Sneath 1963, p. 193).
In the literature, sometimes clustering methods are categorized based on whether the clustering is arithmetic average or centroid clustering, and weighted or unweighted clustering (Sneath and Sokal 1973, p. 228).
7.4.3.3.5 Weighted Versus Unweighted Clustering
Weighted clustering was introduced to weight the individuals unequally, i.e., give merging branches in a dendrogram equal weight regardless of the number of OTUs carried on each branch. Thus, it attempts to give equal importance to phyletic lineages (Sokal and Michener 1958).
Unweighted clustering gives equal weight to each OTU in clusters, evaluates the similarity of OTU with another cluster (or OTU), and thus it less distorts the compared phenograms with original similarity matrices (Sneath 1969) (also see Sneath and Sokal 1973, p. 228).
Weighted method is preferred when very disparate sample sizes are used to represent several taxa. The benefit of using weighted method is when the sparsely represented sample is given more importance, it results in less biased estimates of intercluster distances (Sneath and Sokal 1973, p. 229).
7.4.3.3.6 Arithmetic Average Versus Centroid Clustering
Arithmetic average clustering (Sneath and Sokal 1973, p. 228) “computes the arithmetic average of the similarity (or dissimilarity) coefficients between an OTU candidate for admission and members of an extant cluster, or between the members of two clusters about to fuse.” It does not take into account similarity or dissimilarity (taxonomic distance) coefficients between members within the cluster; thus, it does not consider the density of the points that constitute the extant cluster (or a candidate cluster about to fuse with it) when evaluating the resemblance between the two entities.
Centroid clustering (Sneath and Sokal 1973, p. 228) “finds the centroid of the OTUs forming an extant cluster in an A-space and measures the dissimilarity (usually Euclidean distance) of any candidate OTU or cluster from this point.”
Arithmetic average clustering can be grouped into unweighted pair-group method using arithmetic averages (UPGMA) and weighted pair-group method using arithmetic averages (WPGMA) (Sneath and Sokal 1973, p. 218). UPGMA computes the average similarity or dissimilarity of a candidate OTU to an extant cluster, weighting each OTU in that cluster equally, regardless of its structural subdivision (Sneath and Sokal 1973, p. 230). WPGMA differs from UPGMA in that it weights the member most recently admitted to a cluster equal with all previous members (Sneath and Sokal 1973, p. 234).
Centroid clustering can be grouped into unweighted centroid and weighted centroid (Sneath and Sokal 1973), or just centroid and median (Lance and Williams 1967), respectively. Centroid clustering can also be grouped into unweighted pair-group centroid method (UPGMC) and weighted pair-group centroid method (WPGMC). WPGMC was also called the median method (Lance and Williams 1967). It was first developed by (Gower 1967a). WPGMC weights the most recently admitted OTU in a cluster equally to the previous members of the cluster (Sneath and Sokal 1973, p. 235).
Arithmetic average clustering results in the similar general taxonomic structure as complete linkage analysis, although some fine distinctions exist between them. The disadvantage of arithmetic average clustering is that it does not have easily discernible geometric interpretation as in centroid clustering.
Centroid clustering recognizes the dissimilarities among the points constituting a cluster and hence it has a simple geometric interpretation.
UPGMA is also called group average (Lance and Williams 1967). It works for both dissimilarity and similarity coefficients. It is the most frequently used clustering method in numerical taxonomy and adopted in ecology and microbiome studies. WPGMA has the general taxonomic structure that is identical to UPGMA and shares the properties of UPGMA (Sneath and Sokal 1973, p. 234). But compared to UPGMA, three differences exist in WPGMA (Sneath and Sokal 1973, p. 234): (1) It distorts the overall taxonomic relationships in favor of the most recent arrival within a cluster. (2) In WPGMA the larger clusters are further apart from each other due to the greater dissimilarity in the later joined clusters. (3) The average distances between the clusters are increased because of this heavier weighting in WPGMA.
UPGMC computes the centroid of the OTUs that join to form clusters, and then computes distances or their squares between these centroids, and thus it is attractive conceptually (Sneath and Sokal 1973, pp. 234–235). However, the results of UPGMC procedures are not monotonic due to not meeting the ultrametric inequality (Sneath and Sokal 1973, p. 235). WPGMC shares some properties with UPGMC (i.e., a lack of monotonicity). The heavier weight used to late joiners of clusters causes the taxonomic structure of WPGMC to have some differences from that in UPGMC as implied by the phenogram (Sneath and Sokal 1973, p. 235).
It was shown (Sneath and Sokal 1973, p. 240) by comparing the phenograms from the different clustering methods that the levels of linkage between clusters are on the average highest (least dissimilar) for single linkage and lowest for complete linkage, and intermediate for the average linkage methods. To overcome the disadvantages of single and complete linkage analyses, several modified versions of single and complete linkage clustering have been developed. For example, to avoid the extremes of single linkage (chaining) and complete linkage (small tight compact clusters) clustering, Sneath (1966) proposed four modified linking strategies to link two clusters: (1) integer link linkage, (2) proportional link linkage, (3) absolute resemblance linkage, and (4) relative resemblance linkage. Lance and Williams (1967) proposed a flexible clustering strategy to constrain the linear formula to overcome chaining by single linkage and extremely compact clustering as observed in complete linkage analysis. Shepherd and Willmott (1968) proposed a modified single linkage clustering to prevent chaining. The interested readers are referred to Sneath and Sokal (1973) and references therein for further details of these modified single and complete linkage analyses.
In summary, the average linage clustering method developed by Sokal and Michener has been recommended as clustering method for numerical taxonomy (Sneath and Sokal 1973) because intuitively within-taxon similarity an average measure rather than the minimum or maximum thresholds is more acceptable (Sokal 1985). It can be used as a variable-group method or a pair-group method (Sokal 1963, p. 194). However, using resemblances of taxa to achieve an optimal classification is more complex than maximizing the cophenetic correlation coefficient. Overall criterion should not only consider the average similarity within a taxon and its variance, but also consider the rank of a given taxonomic subset of the entire study (Sokal 1985).
7.4.3.4 Factor Analysis
Taxonomic structure can also be constructed through factor analysis. Based on (Sokal and Sneath 1963), it is Sokal (1958b) who first used factor analysis to analyze taxonomic relationships from a similarity matrix of correlation coefficients, in which factor analysis is used for describing the complex interrelationships among taxa in terms of the smaller number of factors. Here, OTU is used as an observed variable, while taxon is used as a latent factor. The factor loading represents the degree of similarity between an OTU and the average aspect of the taxon. The higher the factor loading, the more typical the OTU belongs to the taxon. Later multiple factor analysis (Harman 1976) was initially used to phenetic classification by (Rohlf and Sokal 1962), in which centroid factor extraction with subsequent rotation is performed to obtain “simple structure” for revealing the interrelationships among OTUs.
Employing factor analysis to classification actually is to define taxa (e.g., species) in terms of similarity of OTUs by using factor analysis. The later bioinformatic work using factor analysis to define species originated in this work of numerical taxonomy. However, the challenge is how many factors should be extracted to define the taxa. In factor analysis the number of factors is usually arbitrarily chosen. Additionally, factor analysis cannot draw straight line across a dendrogram to yield the groups, suggesting that the factor analysis only can approximately, but not exactly form the groups at the same hierarchical level (Rohlf and Sokal 1962; Sokal 1963).
- (1)
It provides important implications for choice of characters. For example, phenetic similarity may be estimated based on the extracted k factors rather than choosing using n characters. In this way such characters are used as necessary to evaluate the factor endowment of each OTU.
- (2)
It theoretically reduces the number of phenetic dimensions necessary to visualize the organisms.
- (3)
It is possible to use factor analysis quickly to find whether more characters should be measured.
However, factor analysis has both practical and theoretical problems. Practically, before a factor analysis is conducted, factor scores and correlations of the characters are needed; thus factor analysis is not really efficient. Additionally, similar as it is a challenge to find clusters of highly correlated characters in cluster analysis, it is even risk to find factors when the characters are highly correlated. Thus, Sneath and Sokal suggested that we might still use the whole set of characters to estimate phenetic similarity from factor scores (Sneath and Sokal 1973, p. 99). Theoretically, it is difficult to know how to weight the extracted factors in measuring the similarity between OTUs. Thus, we need more research to develop an independent criterion for weighting factors.
7.4.3.5 Ordination Methods
Ordinations also present classifications, in which the data are first required to be transformed before being turned into ordinations. The ordination methods in phenetic taxonomy are referred to the methods that describe the occupation of phenetic space by the OTUs being ordinated (Sokal 1986). The ordination methods in application to phenetic taxonomy differentiate from clustering methods, in which OTUs are arranged in a continuous hyperspace with its dimensions being defined by the characters of the OTUs, whereas clustering methods assign OTUs to groups (Sokal and Sneath 1963; Sneath and Sokal 1973; Clifford and Stephenson 1975; Dunn and Everitt 1982; de Queiroz and Good 1997). Both clustering and ordination methods are based on similarities. However, clustering methods aim to base nested hierarchical taxonomies on similarity, whereas ordination methods do not attempt to give hierarchical results, which was reviewed as more accurately representing similarities than do by clustering methods (de Queiroz and Good 1997).
Narrowly OTU-method in numerical taxonomy only includes commonly used clustering methods and factor analysis. Based on (de Queiroz and Good 1997), Sokal pioneered the biological application of ordination techniques, e.g., in (Rohlf and Sokal 1962). However, in their 1963 book, Sokal and Sneath did not include ordination techniques as an approach to phenetic taxonomy. Ordination techniques were described as a potential approach to phenetic taxonomy in their 1973 book (Sneath and Sokal 1973), including principal component analysis (PCA), principal coordinate analysis (PCoA), nonmetric multidimensional scaling (NMDS), and seriation. PCA, PCoA, and NMDS have been readily adopted in microbiome study. We will introduce and illustrate PCA, PCoA, and NMDS as well as other ordination methods with real microbiome data in Chap. 10 (Sect. 10.3). Here, we briefly describe seriation method. For details, the interested readers are referred to Sneath and Sokal (1973) and Legendre and Legendre (2012) and the references therein.
Seriation is a simple type of (one-dimensional) ordination previously developed in anthropology (Petrie 1899; Czekanowski 1909) and archaeology (Craytor and Johnson Jr 1968; Johnson Jr 1968; Kendall 1971, 1988). It was first applied to ecology by (Kulczynski 1927). Seriation in archaeology (Hodson 1970; Cowgill 1968) and ecology (Legendre and Legendre 2012) is used as a clustering analysis.
Seriation is to permute the rows and columns of a similarity matrix in such a way as to arrange the highest similarities (or the lowest distances) near the principal diagonal of the similarity (resemblance) matrix and with the similarity values decreasing orderly away from the diagonal. Thus, seriation method essentially is how to operationally define order among the coefficients in a similarity matrix (Sneath and Sokal 1973, pp. 250–251).
The seriation method has been reviewed having some shortcomings (Sneath and Sokal 1973, p. 251). For example, (1) this technique requires an iterative algorithm to order all possible arrangements of OTUs and hence is computationally expensive (Craytor and Johnson Jr 1968); (2) it is prone to be local optima (Kendall 1963; Craytor and Johnson Jr 1968); (3) it is only applicable to very specialized data sets such as in cases of true linear trend development; (4) it has been reviewed that seriation is inferior to NMDS and principal axis methods (Cowgill 1968; Kendall 1969; Hodson 1970), as well as less specific and efficient than the clustering methods in analysis of a usual similarity matrix since seriation does not directly generate clusters of objects (Legendre and Legendre 1998, p. 372).
Seriation also has the attractive property because it can be applied to both non-symmetric and symmetric similarity or distance matrices (Legendre and Legendre 2012, p. 403). This interesting property may attract the microbiome researchers to apply this method in their studies since as in ecology, the similarity or distance matrices in microbiome may be not symmetric. The R package seriation has been developed for performing seriation analysis (Hahsler et al. 2008).
7.4.3.6 Discriminant Analysis in Identifying OTUs
Numerical taxonomy makes distinction between classification and identification. As we described in Sect. 7.2.3, at the beginning, unequal weighting was not promoted in numerical taxonomy. However, Sokal and Sneath recognized that strictly equal weighting is impossible in their later publications, such as in (Sneath and Sokal 1973). They distinguished identification from classification, in which identification or assignment is used to assign a new entity (Dagnelie 1966).
Classification is used in the sense of making classes, clusters, or taxa. By contrast, identification is used for “identifying” new individuals (OTUs/taxa). However, when we identify an individual as new one, we actually know that an existing class is unacceptable. The cluster analyses can effectively work on the classification. But to identify new individuals, it requires that some strategies can combine the two procedures of classification and identification. The discriminant functions (analyses) are the main methods used in identification.
Along with the distinction between classification and identification, Sneath and Sokal believed that weighting is needed. The goal of weighting characters in numerical taxonomy is for identification. Sneath and Sokal (1973, p. 400) thought that weighting can maximize the probability of correctly identifying unknown specimens from a few close or overlapping taxa through a set of multivariate statistical methods.
Sneath and Sokal have specifically introduced three discriminant methods in describing or discriminate OTUs (or taxa): discriminant function analysis (DFA), and multiple discriminant analysis in Mahalanobis’ D-space and canonical variates analysis. We can view these multivariate statistical methods as extensions of taxon distance models in Sect. 7.4.2.3. The purpose of taxon distance models in Sect. 7.4.2.3 is to classify the OTUs, while the purpose of discriminant analyses is to perform both classification and identification, i.e., to discriminate the OTUs (or taxa).
7.4.3.6.1 Discriminant Function Analysis (DFA)
DFA or linear discriminant analysis (LDA) or linear discriminant function analysis (LDFA) is a generalization of Fisher’s linear discriminant method (Fisher 1936). LDFA is a member of the family of classification methods known as supervised learning, in which group membership of individuals is used to maximize group differences during the process. In this method, a linear combination of features that characterizes or separates two or more classes of objects is to be identified and to be used as a linear classifier or for dimensionality reduction before later classification. LDA requires that groups (i.e., the class label) are known a priori.
In numerical taxonomy, the linear discriminant function is a linear function z of characters describing OTUs that weight and separate the characters in such a way to identify two taxa: maximizing number of the OTUs in one taxon has high values for z and maximizing number of the OTUs in another taxon has low values, so that z serves as a much better discriminant of the two taxa than does any one character taken singly (Sneath and Sokal 1973, p. 400). By performing the linear discriminant function, (1) the n characters are almost always reduced to a smaller set, m; and (2) the function also has maximal variance between groups relative to the pooled variance within groups.
Step 1: The linear discriminant function generates a set of taxa (i.e., three taxa J, K, and L) for all the character axes (i.e., X1 and X2). The generated taxa are represented by the clusters of individuals (OTUs).
Step 2: After clustering individuals (OTUs), we now only concern the descriptive parameters of the clusters (i.e., three taxa J, K, and L), i.e., the centroids and the dispersions of the clusters and do not concern with the values for the individuals.
Step 3: First calculate the variances and covariances between the characters, yielding a set of (here three) m × m matrices (here m = 2); then average these variances and covariances to give a pooled within-groups variance-covariance matrix W.
Step 4: Calculate the discriminant function between J and K. Multiply the inverted W matrix by the vector
![$$ {\delta}_{JK}=\left[\left({\overline{X}}_{1J}-{\overline{X}}_{1K}\right),\left({\overline{X}}_{2J}-{\overline{X}}_{2K}\right),\dots, \left({\overline{X}}_{mJ}-{\overline{X}}_{mK}\right)\right] $$](images/486056_1_En_7_Chapter_TeX_IEq23.png) to obtain the discriminant function as a vector z: zJK = W−1δJK. This vector consists of a series of weights, wi, for characters 1, 2, …, m, it is symbolized as z1, z2, …, zm.
to obtain the discriminant function as a vector z: zJK = W−1δJK. This vector consists of a series of weights, wi, for characters 1, 2, …, m, it is symbolized as z1, z2, …, zm.Step 5: Calculate a discriminant score DS (here the discriminant score scale for taxa J and K). Multiply these weights by the observed character values of the unknown individual, and sum them to give a discriminant score DS: DSu = z1X1u + z2X2u + … + zmXmu.
Step 6: Use this discriminant score to calculate three reference scores for the centroid of J, the centroid of K, and the point midway between them: DSJ, DSK, and DS0.5 respectively for discriminating between members of J and K. They are given by:
Step 7: Calculate the length of the line between the centroids of J and K and allocate the unknown to taxon. First, obtain the square root of Mahalanobis’ D2, which is the length of the line between the centroids of J and K measured in discriminant function units (the absolute difference between the scores DSJ and DSK is also equal to D2); then allocate the unknown to taxon J if the observed score for an unknown,DSu, lies on the DSJ side, otherwise to taxon K if on the D DSK side. A different discriminant function, and discriminant scores, is calculated for each pair of taxa. The square root of Mahalanobis’ D2 (Mahalanobis 1936) is used to measure the length of the line between the centroids of two taxa J and K in discriminant function units, which equals to the absolute difference between the discriminant scores of these two taxa DSJ and DSK.
Step 8: Interpret discriminant function vectors. The vector angle between two discriminant function vectors can be interpreted as measuring contrasts of form of the taxa, where the small angle measures similar contrasts of form and large angles represent distinct contrasts.
Step 9: Perform statistical hypothesis testing of the centroids using the distance scores. The distance
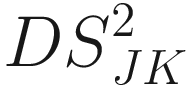 between the scores DSJ and DSK (Sneath and Sokal 1973, pp. 403–404) has two important uses. One is that we can perform a null statistical hypothesis through a F-test to test whether the centroids are significantly different with a testing ratio:
between the scores DSJ and DSK (Sneath and Sokal 1973, pp. 403–404) has two important uses. One is that we can perform a null statistical hypothesis through a F-test to test whether the centroids are significantly different with a testing ratio: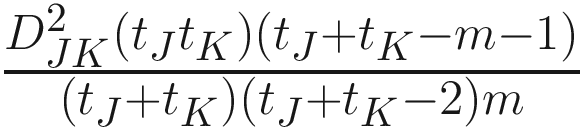 , with m and (tJ + tK − m − 1) degrees of freedom.
, with m and (tJ + tK − m − 1) degrees of freedom.
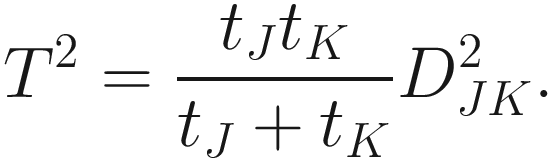
Step 10: Perform variable (character) selection. We also can use the distance scores to determine each character contributing to D2. If the characters with little discriminatory power can be identified, then drop them from the analysis. Alternatively, we can choose the best few characters from the set by the F test. If we do not consider correlations between characters, the percent contribution of character i is 100 × (ziδi/D2), where zi and δi are the ith elements of vectors zJK and δJK.
Step 11: Calculate the probability of misclassification. A primary purpose of a discriminant function is to minimize the probability of wrong assignment of unknown individuals. We can calculate the probability of misclassification of assignment of unknown individuals by assuming a multivariate normal distribution and also that the unknown does belong to J or K (and not to some distant cluster).
7.4.3.6.2 Multiple Discriminant Analysis (MDA)
The original dichotomous discriminant function has two limitations (Sneath and Sokal 1973, p. 404): (1) The algorithm used in the original dichotomous discriminant function implies that the unknown either belongs to one or the other of the two taxa being considered. Thus, when an unknown taxon is located far off in the space, belongs to a quite different cluster, and hence it really belongs to a third cluster. However, using the dichotomous discriminant function, this unknown taxon may have almost any discriminant score, and thus is assigned to one or the other of the two taxa under consideration. (2) It cannot test against a large number of discriminant functions when there are many taxa.
MDA in Mahalanobis’ D-space (Mahalanobis 1936) was developed to largely overcome the above two problems of the original dichotomous DFA. MDA is a multivariate dimensionality reduction technique. It is not directly used to perform classification, instead merely supports classification by compressing the signal down to a lower-dimensional space amenable to classification. MDA typically projects the multivariate signal down to a dimensional space with less one dimension of the number of categories. Mahalanobis (1936) generalized calculation of D2 between the centroids from the discriminant scores to between any pair of points so that the square root of D2 become simply the Euclidean distance in the D-space. The Mahalanobis’ D2 between any pair of points f and g is given:
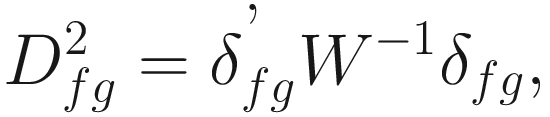
MDA method transforms the original space into a new space, where the original axes are stretched and also skewed, and results in not any more right angles between axes.
The length of a unit in dimension equals to the original units times by the square root of the element of the matrix w−1 (Sneath and Sokal 1973, p. 405). The direction cosine between the dimensions has an interpretation of correlation coefficients (Gower 1967b).
7.4.3.6.3 Canonical Variate Analysis (CVA)
CVA is a widely used method for analyzing group structure in multivariate data. It is mathematically equivalent to a one-way MANOVA (multivariate analysis of variance) and often called canonical discriminant analysis. The reason that we can perform analyses on distances between taxa or individuals, in particular by principal coordinate analysis is that the square root of D (the Mahalanobis’ D2), the difference between discriminant scores of two taxa can be represented in an orthogonal system of axes (though the orientation is arbitrary) (Gower 1966, 1967b) (also see (Sneath and Sokal 1973, p. 406)).
CVA and MDA are equivalent except in minor particulars. We can obtain the coordinates of the points in an orthogonal system such as by principal coordinate analysis of D, and the orthogonal axes are the canonical variates, which can be used also for discrimination to readily identify unknowns by seeing whether they fall within critical distances of taxon centroids.
DFA and the Mahalanobis’ D2 have the advantages: (1) they are less sensitive to general size factors than taxonomic distance (Sneath and Sokal 1973, p. 406), although an unknown of the same shape as a member of a taxon may appear outside that taxon if it differs much in size. (2) Discriminant functions have most value for very close clusters that partly overlap (Sneath and Sokal 1973, pp. 406–407).
- (1)
The pooled within-groups variance-covariance matrix W cannot be inverted, unless “generalized inverses” are used if any character is invariant in each of the taxa.
- (2)
It is difficult to choose the limited set of the best characters from the large number, which should be employed in numerical taxonomy. Characters with means that are well separated in relation to the variances and that are not highly correlated with other characters are in general the best, but optimal methods of choosing them pose statistical and computational problems (Feldman et al. 1969).
- (3)
It has been shown by Dunn and Varady (1966) that rather large numbers of individuals are required in each taxon for reliable discriminant functions.
- (4)
The gain in discriminatory power over simpler methods may not be great (Sokal 1965) particularly with 0, l characters (Gilbert 1968; Kurczynski 1970) and simple discriminants based on equal weight for each character can be quite effective (e.g., Kim et al. 1966).
7.5 Statistical Hypothesis Testing of OTUs
Statistical hypothesis testing of OTUs is challenging in performing both statistical inference of similarity coefficients and clustering.
7.5.1 Hypothesis Testing on Similarity Coefficients of OTUs
Numerical taxonomy concerns more on general significance of the similarity matrix among all the OTUs or the joint significance of the entire matrix. In numerical taxonomy, it is not important to conduct statistical hypothesis testing significance of each individual similarity coefficient. There are two reasons behind this (Sokal 1963, pp. 153–154). We briefly describe below.
7.5.1.1 Heterogeneity of the Column Vectors of the Data Matrix
In the OTU Data Matrix, the similarity between OTUs j and k are measured over a set of n characters. The problem is that these OTUs are not taken as random samples from a common population, but instead are really taken from a heterogeneous sample wherein each variate estimates a different character. In other words, the character states for any one OTU are not samples that estimate a single mean and variance. Thus, because the column vectors of the data matrix are heterogeneous, it is difficult to assume the sampling parametric distributions for justifying statistical test; and hence it is inappropriate to make ordinary tests of significance (Sokal and Sneath 1963, p. 313). Since a parametric model cannot be easily specified, in ecology and microbiome study, a permutation test is often used to perform a hypothesis testing.
7.5.1.2 Hypothesis Testing of Individual Similarity Coefficients
An appropriate parametric statistical test in multivariate statistics requires the variables (in this case, characters) are independent so that there are p variables (characters) in the data matrix, and then the data matrix has p dimension with each of the p variables representing one dimension of the data. The problem is that in OTU data matrix, the characters (rows of the data matrix) are correlated with each other, and thus a similarity coefficient based on p characters would not reflect p independent dimensions of variation (Sneath and Sokal 1973, p. 163). Thus, standardization of characters is often recommended in numerical taxonomy to alleviate the heterogeneity of column vectors. However, standardization of every character state to have a population mean of zero and variance of unity only mitigate some degree of heterogeneity of column vectors. Actually, put another way, statistical hypothesis tests in numerical taxonomy only have partially succeeded for entire classifications (similarity matrices), but not for individual coefficients (Sneath and Sokal 1973, pp. 163–164). In numerical taxonomy, scaling continuous characters (variables)(lengths, percentages, ratios) is recommended by either standardization or ranging scaling methods. Because of scaling characters, correlation or taxonomic distance matrices between OTUs hence are altered. Two states of characters should be coded as binary, while ordered discrete multistate characters (qualitative traits and counts) can be analyzed as continuous characters (Sokal 1986).
These two problems actually reveal the challenges of directly conducting the hypothesis testing of individual similarity coefficients in numerical taxonomy, and ecology as well as in later microbiome study. Performing hypothesis testing pairwise correlation or partial correlation also poses a challenge. In a similar reason, beta diversity measures such as Bray-Curtis, Jaccard, and Sørensen dissimilarities are usually used as input data for ordination and association analysis rather than directly being tested. This links up the concepts and topics in multivariate statistics, with which we will discuss in Chapters 10 and 11.
7.5.2 Hypothesis Testing on Clustering OTUs
One critical step in numerical taxonomy is to assemble the OTUs into groups of higher rank using the similarity coefficients. In other words, the taxonomic groups are formatted by using cluster analysis (or multiple factor analysis, which is somewhat similar to cluster analysis) using the similarity coefficients as criteria of rank (Sneath and Sokal 1962; Sokal and Sneath 1963). As we described above, it is because the OTUs (or taxonomic groups) are formed using cluster analysis, we call the OTU methods as clustering-based OTU methods. However, it is also difficult to perform hypothesis testing on clustering OTUs. The cluster analytic procedures of numerical taxonomy have been frequently criticized due to lack of a sound basis in statistical inference (Cormack 1971; Sokal 1986).
Several different approaches of significance tests for clusters have been comprehensively reviewed and discussed in literature (Dubes and Jain 1979; Bock 1981, 1985, 1996, 2002, 2012; Perruchet 1983).
Generally it is not recommended to globally apply a cluster test to a large or high-dimensional data set; however, performing a “local” cluster test (e.g., the maximum F test with 2 classes) to a specified part of the data is often useful for providing evidence for or against a prospective clustering tendency (Bock 1985).
Most cluster analyses test the null hypothesis that there is no structure at all such as test for hierarchical structure versus random data (Rohlf and Fisher 1968). However, this test of clustering is less important in biological classification because it is most likely that the organisms representing a taxonomic group must have taxonomic structure (Sokal 1986). The advances of clustering methods from classical models to the approaches developed until 2002 was reviewed by Bock (2002).
To address the problem of assessing the validity of clusters produced by classical clustering methods, some advances were made in testing for significant clusters, including permutation test for significance (Good 1982; Park et al. 2009) and Monte Carlo test for assessing the value of a U-statistic based on the sets of pairwise dissimilarities to identify genuine clusters in a classification (Gordon 1994).
However, probabilistic clustering models for noisy and outlying data (Bock 2002, 2012) may remedy some degrees of limitation of classical clustering methods. Probabilistic clustering models were developed for testing for homogeneity versus clustering; that is, to test the discriminating (distinguishing) of homogeneity and heterogeneity in multidimensional observations. Typically statistical methods in probabilistic models discriminate whether the observations are sampled from “homogeneous” population (the null hypothesis) or from “heterogeneous” (or “clustering”) population (the alternative hypothesis). Bock (2002) reviewed and discussed two model-based probabilistic clustering models which lead to a new “robust” clustering method: (1) modeling noisy data in clustering (Banfield and Raftery 1993); (2) modeling outliers and robust clustering (Gallegos 2001, 2002, 2003).
Summary statistics are commonly used in various fields of research. Like in numerical taxonomy, currently in microbiome study, the techniques of summary statistics, such as alpha and beta diversity measures, ordination, correlation, and partial correlation analyses, and clustering are all used to summarize data and measure taxonomic structure.
However, clustering in numerical taxonomy is based on phenetic resemblances (affinity) only without considering phyletic connotations (Sokal 1963, p. 52). The question still remains to be asked: (1) how it is appropriate to adopt similarity coefficients and clustering of OTUs into microbiome data? (2) how the statistical hypothesis testing of similarity coefficients and clustering of OTUs is clinically relevant? We feel that overall test of similarity or dissimilarity structure using clustering methods does not provide much meaningful clinical insights in current microbiome study. Further functional studies in experimental models and validation using different cohorts are needed.
7.6 Some Characteristics of Clustering-Based OTU Methods
Describing the main characteristics of numerical taxonomy and especially major characteristics of clustering-based OTU methods will not only facilitate understanding the field of numerical taxonomy per se, but also help understanding the advantages and limitations of the concept of OTUs and the clustering-based OTU methods that were adopted in microbiome study. In this section, we summarize some basic questions and the main characteristics of numerical taxonomy and especially of clustering-based OTU methods.
7.6.1 Some Basic Questions in Numerical Taxonomy
Numerical taxonomy has provided its unique resolutions to some basic questions in taxonomy or classification in systematics. We describe the main ones below.
Sokal and Sneath definitely stated that no absolute evolutionary rates exist, and all groups are ranked based on investigating and evaluating its characters and phenetic resemblances and thus evolutionary rates are related to the higher ranking groups and to neighboring taxa (Sokal 1963, p. 239). For them, the affinity values provided by the techniques of numerical taxonomy can be used to measure the evolutionary rate (Sokal 1963, p. 241).
Whether phenetic similarity is concordant with taxonomic species, and whether there exist the correlation (correspondence) between genetic relationships and phenetic groupings are debatable.
Some researchers thought that a very close correspondence between them cannot be found, while others thought that they could find reasonable agreement between phenetic relations and the ease of hybridization. Sokal and Sneath emphasized that there exists a sharp distinction between phenetic grouping and genetic groupings (Sokal and Sneath 1963, p. 243). They thought that phenetic, genetic, and geographical groupings cannot always be equated each other. Thus, two distinct genetic groups would be within a single phenetic group. However, they believed that phonetically distinct groups identified by numerical taxonomy will usually be found to be genetically distinct.
If a pattern of taxonomic dendrograms occurs at random, then the number of side branches per stem in a given interval is distributed with a Poisson distribution with the number of OTUs being the total number of side branches plus one. Obviously, a Poisson distribution is not appropriate to describe the pattern of branching of taxonomic dendrograms and the phenetic distributions. In taxonomic dendrograms, given a Poisson distribution, the variance is much greater than the mean number of branches per phenon, and the observed number with no branches (monotypic phenons) is greater than the expected [see (Sokal and Sneath 1963, pp. 245–246; Sneath and Sokal 1973, p. 307)]. Sokal and Sneath (1963, p. 246) called this kind of over-dispersed and zero-inflated distribution as a clustered distribution and thought that it has affected on the construction of hierarchies. In their 1973 book (Sneath and Sokal 1973, p. 308), they further described three properties of phenetic distributions: (1) there exist some dense clusters of lower taxa; (2) there exist a great number of isolated monotypic taxa; and (3) some constancy exists in the organized nature or in the constructed rank levels which is indicated by the consistency of the slope of the logarithm of the number of included taxa.
7.6.2 Characteristics of Numerical Taxonomy
In this subsection, we describe some characteristics of numerical taxonomy.
7.6.2.1 Summary of Characteristics of Numerical Taxonomy
- 1.
Construction of classifications does not necessarily require phylogeny.
In numerical taxonomy classifications are called nature or “general” classifications, and the constructed taxa have “natural” properties or are “natural” taxa/“natural taxa.” For Sneath and Sokal (1962), “natural taxonomic groups” are “polytypic” (Beckner 1959) or better called as “polythetic” (meaning “many-arrangement”), which is against “monothetic” (meaning “one-arrangement”).
Sokal and Sneath tried to avoid confusing phylogeny with classfication (Huxley 1869) and emphasized that taxonomic classifications are distinctive from phylogenetic classifications. Basically numerical taxonomy does not attempt to force phylogenetic criteria on to taxonomic groupings. Sokal and Sneath separated the taxonomic process (which is to be based on affinity or resemblance) from phylogenetic speculation (Sneath and Sokal 1962). They emphasized that taxonomic relationships are “static” (Michener 1957) or preferably are called “phenetic” (Sneath and Sokal 1962; Cain and Harrison 1960), which are to be evaluated purely based on the resemblance among the materials. In numerical taxonomy it is logically fallacious to make phylogenetics deductions to arriving at taxonomies. Sokal and Sneath descibed a fallacy of circular reasoning (Sokal and Sneath 1963, p. 21):
“Since we have only an infinitesimal portion of phylogenetic theory in the fossil record, it is almost impossible to establish nature taxa on a phylogenetic basis. Conversely, it is unsound to derive a definitive phylogeny from a tentative natural classification.”
- 2.
Natural taxonomic classifications should be constructed based on similarity coefficients using many characters.
In numerical taxonomy, taxa are based on correlations between features (Sneath and Sokal 1962), and taxonomic rank is defined by similarity (Sneath 1957a). The phenetic resemblance between the taxonomic entities is estimated using the similarity and dissimilarity coefficients (Sneath and Sokal 1962).
Three convenient types of coefficients (association, correlation, and distance) were suggested in numerical taxonomy as suitable measures of similarity between pairs of taxa (Sokal 1961). Specifically association coefficient is utilized to measure “present-absent” (binary) characters, which was first described by Sneath (Sneath 1957a). Binary data have attractive properties of simplicity and the potential to decompose variation among OTUs into fundamental units of information. The simple matching coefficient (including its one-complement coefficient, the average Manhattan distance coefficient) and the Jaccard coefficient were recommended for use in numerical taxonomy among the many binary association coefficients that most have been used in ecology (Sokal and Sneath 1963; Sokal 1986).
The ordinary Pearson product-moment correlation coefficient is still preferred to use as measures of similarity between OTUs because it is less sensitive to size differences, although it is non-metrics and non-invariant to the direction (Sokal 1986). In contrast, cosine coefficient is metrics and invariant to the direction of correlation coefficients. For continuous or ordered multistate characters (variables), both taxonomic distances and Manhattan distances can be used to calculate similarity and dissimilarity. Distance coefficients determine the similarities between two taxa as a function of their distance in a multiple dimensional space with the characters as coordinates. They are employed to measure continuous or more than two states (ordered multistate) of the characters (variables) (Sokal 1958a; Morishima and Oka 1960). Between taxonomic distances and Manhattan distances that can calculate similarity and dissimilarity, Sokal (1986) preferred taxonomic distances because of their geometric and heuristic properties. Based on Sokal (1961), the use of distance to measure taxonomic similarity can be located earliest by Heincke (1898) and others (Anderson and Abbe 1934; Pearson 1926; Rao 1952; Mahalanobis 1936). Both correlation and cosine coefficients are sensitive to data points close to the centroid. However, classifications in numerical taxonomy are preferred to be based on similarity using correlation (or cosine) coefficients than by using distance-based measures because the former usually provide better effects of classifications to exhibit size and shape differences in the data (Sokal and Rohlf 1980; Sokal 1986).
- 3.
Characters should be equally weighted.
- 4.
Taxonomy approach is quantitative and numerical.
Taxonomic groups are numerically established by cluster analysis of values of similarity. To exploit the taxonomic structure, numerical taxonomy uses different clustering methods to group OTUs into clusters for achieving the taxonomic structure. Three commonly used clustering methods in numerical taxonomy are single linkage clustering, complete linkage clustering, and average linkage clustering methods. Among these three clustering methods, the average-linkage clustering method was promoted; and particularly the unweighted pair-group method using arithmetic averages (UPGMA) was also preferred for use because the average clustering method can present a compromise solution to form a cluster (Sokal 1986).
It is also because single-linkage and complete-linkage clustering methods represent the two extreme criteria and both could not give taxonomically satisfactory results (Hartigan 1985), although single linkage clustering has several elegant mathematical properties (Sokal 1986). Thus they both are used in special cases only (Sokal 1963; Sneath and Sokal 1973).
- 5.
The concept of operational taxonomic units (OTUs) is purely “operational,” and thus it is not biologically motivated.
OTUs were introduced as a pragmatic definition to classify groups of organisms, allowing for taxonomy being classified quantitatively or numerically. Thus, the concept of OTUs and the field of numerical taxonomy were established against the subjective nature of classification in classical Linnaean taxonomy. Through creating the concept of OTUs, the term “taxon” can still be retained for its proper function role played in taxonomic groupings. Thus, this indicates that OTUs are not true taxa instead are operated as taxa. OTUs are used in constructing classifications without considering any phylogenetic information. Actually by using the OTUs, the classification was considered purely as a study of similarity, of “static” relationships in which phylogeny played no part (Vernon 1988). Thus, when constructing phylogenetic classifications, the static relationships and phylogeny should be separately assessed.
7.6.2.2 Remarks on Characteristics of Numerical Taxonomy
The approach of phonetic numerical taxonomy (or phenetics) and commonly clustering-based OTU methods have several challenges.
First, phonetic numerical taxonomy (Gilmour naturalness) assumes that high similarity (in fact largely clustered) will also lead to high predictivity. This has been identified as an unfinished problem (Farris 1979; Sokal 1985) because the linkage between similarity and predictivity has not been defined clearly. Actually, on one hand, it is challenging to show how to maximize similarity; on the other hand, the relationship between classification and predictivity has not yet been established although a close relation has obviously been observed between classifications in which objects within any given taxon resemble each other closely and predictivity of characters within that taxon (Sokal 1985). Predictivity is the most important criterion of a classification in terms of Gilmour naturalness. Thus, although it is difficult to achieve, generally high predictivity is the goal of a natural classification (Sokal 1985).
For example, phenetic clustering is commonly used in specifically and biologically estimating phylogeny or phylogeny reconstruction, and is also used to analyze relationships among potentially conspecific populations, as well as geographic variation and genetic continuity, and consequently, of species limits (Highton et al. 1989). Both above applications of phenetic clustering have been criticized having serious limitations because of their assumptions are questionable (de Queiroz and Good 1997). When phenetic clustering is used for estimating phylogeny, it assumes that similarity is correspondent to recency of common ancestry, while when it is used for analyzing patterns of geographic variation and genetic continuity among populations, it assumes that similarity is correspondent to degree of genetic continuity.
The fundamental weakness of numerical taxonomy and hence its clustering-based OTU methods is its questionable assumptions. Because similarity relationships do not strictly conform to the pattern of a nested hierarchy, and thus attempts to base nested hierarchical taxonomies on similarity will never be fully satisfactory (de Queiroz and Good 1997).
Second, the classifications in biological taxonomy have the property that the resulting classification should be hierarchical and nonoverlapping; and hence requires the resulting classification represents faithfully as possible the actual similarities among all pairs of OTUs resemblance matrix, and the measures (e.g., the cophenetic correlation coefficient) can express the goodness of a classification (Sokal and Rohlf 1962; Sokal 1985). However, it was shown that different clustering methods have different goodness of fits by cophenetic correlation coefficients to the same resemblance matrix (Sokal 1985). Both artificial data (Sneath 1966) and real data (Sokal 1985) have shown average-linkage clustering outperformed complete and single linkage clustering methods to measure the resemblances.
Overall, in one hand, importantly phenetic clustering OTUs expects to result in nested hierarchical structure. However, similarity that does not exhibit a strictly nested hierarchical structure has important consequences for its analysis using phenetic clustering (de Queiroz and Good 1997). On the other hand, even random distributions of OTUs will result in the formation of clusters [see(Sneath and Sokal 1973, p. 252; Dunn and Everitt 1982, p. 94; de Queiroz and Good 1997)].
Third, whether clustering methods or ordination analysis methods better represent taxonomic structure is still arguable. The phenetic criteria of goodness of classification are restricted to hierarchic Linnaean classifications. However, non-Linnaean modes of data analysis would provide better fits to the original resemblance matrix than dendrograms generated by hierarchic cluster analysis methods; thus, ordinations could summarize the taxonomic structure, and hence various ordination analysis methods have been employed in phenetic taxonomy; on the other hand, the hierarchic clustering analysis models have been used as standard methods for classification in the narrow sense (Sokal 1985). Given robust methods for similarity coefficients, Sokal (1985) considered that cluster analysis may ultimately result in obtaining stable and repeatable phenetic classification although it is not possible to achieve complete stability.
7.6.2.3 Remarks on Impact of Numerical Taxonomy on Microbiome Research
The philosophy, OTU concept, framework of statistical analysis of phonetic numerical taxonomy, and especially commonly clustering-based OTU methods have been adopted into microbiome research. All these have provided a solid foundation in the development of microbiome research, especially in the theoretical and conceptual frameworks. The philosophy and methodology of numerical taxonomic procedure have posed large impacts on early microbiome study. However, the limitations and challenges of phonetic numerical taxonomy both as a method for clustering OTUs, and as a statistical analysis method for assessing taxonomies and microbiomes still remain, and even more severe when using OTU methods to define species.
First, OTU concept and its clustering-based OTU method. This may be one of the major impacts of numerical taxonomy on microbiome study. This has been especially reflected in the early development of bioinformatic software such as in the open source software package mothur for bioinformatics data processing.
Second, similarity and distance measurements. Various approaches of grouping OTUs into different levels of taxonomy in early bioinformatic analysis of microbiome sequencing data are just using the algorithms that are related to similarity or distance measurements developed in numerical taxonomy.
Third, ordination and discriminate analysis. Using ordination analysis to represent taxonomic structure and discriminant analysis to classify and identify OTUs (or taxa) provide meaningful insights into current microbiome study. For example, in Sneath and Sokal 1973 book, they made distinction between clustering and ordination, and discussed the advantages of ordination over clustering. They also made distinction between classification and identification, and described several discriminant analysis methods in the role of identifying new taxa. These contributions should be acknowledged.
Forth, correlation between clusters of OTUs and taxa. Overall correlated clusters of OTUs empirically observed by numerical taxonomic methodology may suggest a causal explanation or a causally associated with some mechanisms.
However, both philosophy and methodology of numerical taxonomy have their own fundamental weaknesses. When the philosophy and methodology of numerical taxonomy, and especially the clustering-based OTU methods, are adopted into microbiome study to define species, these weaknesses have not disappeared but somehow have been highlighted.
It confuses the phenetic species with the biological species (or used by microbiologists). Sokal and Crovello (1970) criticized the biological species concept because the concept of biological species is based on modern evolutionary theory using the population as its basic unit, and it is considerably difficult to define a species. They emphasized that the biological criteria for species should be dependent on phenetic considerations. In their 1973 book (Sneath and Sokal 1973, pp. 362–367), Sneath and Sokal discussed various species concepts and definitions and especially described three criteria that are used to define three major species concepts: genetic, phenetic, and nomenclatural. They are respectively referred to (1) genospecies (a group of organisms exchanging genes), (2) taxospecies (a phenetic cluster), and (3) nomenspecies (organisms bearing the same binomen) in Ravin (1963).
Sneath and Sokal particularly discussed the biological species concept that was foremost advocated and defined by Ernst Mayr (1963), in which a biological species is defined as comprising “groups of actually or potentially interbreeding populations which are reproductively isolated from other such groups.” However, they had no intention of using their OTU methods to define a biological species; instead they (Sneath and Sokal 1973, p. 364) explicitly stated that the OUT method defines the phenetic species but not the biological species and that the biological species concept is in practice nonoperational (Sokal and Crovello 1970) and is misleading because the species described by conventional phenetic criteria does not have the genetic properties ascribed to the biological species (Blackwelder 1962; Sokal 1962).
The methodology and especially the clustering based-OTU methods in numerical taxonomy have played an important role in defining OTUs in bioinformatic analysis of microbiome. In the meantime, we want to emphasize that both the philosophy and methodology of numerical taxonomic procedure have their limitations. When the clustering based-OTU methods are used in microbiome study to define species, tasks have been actually assigned: not only for construction of taxonomy but also implicitly requiring for a biological or functional role that the defined species should have. This is beyond the original means described in numerical taxonomy.
Numerical taxonomy along with its clustering based-OTU methods was a reform movement that arose in response to a dissatisfaction with the subjective and nonquantitative nature of taxonomy as it existed in the late 1950s (Vernon 1988). However, the applications of phenetic clustering methods within the context of taxonomy and in the biological sciences have never been entirely satisfactory (de Queiroz and Good 1997). The application of clustering based-OTU methods to bioinformatic classification of OTUs in the field of microbiome still neither is entirely satisfactory.
Actually, a reform movement in response to a dissatisfaction using clustering based-OTU methods to define taxonomy that has already started in the bioinformatic analysis of microbiome sequences. We will comprehensively review the bioinformatic analysis methods and the movement beyond OTUs in Chap. 8.
Numerical taxonomy in its original setting mainly is a purely descriptive science; its potential to be used for formulating hypotheses has not been fully developed in numerical taxonomy. The potential for formulating hypotheses is developed by microbiome researchers in their differential abundance analysis and association analysis. We will cover statistical analysis methods and models in Chaps. 9, 10, 11, 12, 13, 14, 15, 16, 17 and 18.
7.7 Summary
In this chapter we described the original OTU methods in numerical taxonomy.
First, the brief history of numerical taxonomy was introduced. Then, the principles of numerical taxonomy were described, including definitions of characters and taxa, sample size calculation, and equally weighting characters and taxonomic rank. Followed briefly introduced phenetic taxonomy, the philosophy of numerical taxonomy, and methodology, including the numerical-based empiricism, the numerical-based empiricist natural classification, and biological classifications. We focused on how to construct taxonomic structure, which includes defining the operational taxonomic units (OTUs), estimating taxonomic resemblance, commonly clustering-based OTU methods, factor analysis, ordination methods, discriminant analysis, and canonical variate analysis. Particularly, the advantages and disadvantages of single linkage, complete linkage, and average linkage clustering were described and discussed. Next, the problems of statistical hypothesis testing of OTUs using similarity coefficients and clustering methods were described and discussed. Finally, some characteristics of clustering-based OTU methods and their applications to microbiome research were investigated and commended. In Chap. 8, we will introduce and describe the movement of moving beyond the concept of OTUs and OTU methods in bioinformatic analysis of microbiome data and generally in microbiome study.