In Chap. 7 we introduced clustering-based OTU methods in numerical taxonomy. In this chapter we will discuss the movement of moving beyond OTU methods and investigate the necessity and possibility of this movement. We divide the presentation into four sections: We first describe clustering-based OTU methods and the purposes of using OTUs and definitions of species and species-level analysis in microbiome studies (Sect. 8.1). We then introduce the OTU-based methods that move toward single-nucleotide resolution (Sect. 8.2). Section 8.3 describes moving beyond the OTU methods and discusses the limitations of OTU methods. Section 8.4 describes the movement of moving beyond OTU methods and discusses the necessity and possibility of moving beyond OTU methods. Finally, we summarize the contents of this chapter (Sect. 8.5).
8.1 Clustering-Based OTU Methods in Microbiome Study
In microbiome studies, there exist two widely used approaches of sequence similarity: clustering-based OTU methods and phylotype-based (phylotyping) methods. Both approaches use similarity measure and group sequences into small units, which are rooted in clustering methods in the original numerical taxonomy.
8.1.1 Common Clustering-Based OTU Methods
Clustering-based OTU methods typically first perform a hierarchical cluster analysis and then assign the sequence reads into OTUs based on a distance threshold, i.e., cluster their similarity to other sequences in the community (Schloss and Westcott 2011; Huse et al. 2010; Schloss and Handelsman 2005; Schloss et al. 2009; Sun et al. 2009).
In contrast, phylotype-based methods classify sequences into taxonomic bins (i.e., phylotypes) based on their similarity to reference sequences (Schloss and Westcott 2011; Huse et al. 2008; Liu et al. 2008; Stackebrandt and Goebel 1994). For example, taxonomy-supervised analysis (Sul et al. 2011) belongs to the phylotyping methods, in which sequences are allocated into taxonomy-supervised “taxonomy bins” to provide 16S rRNA gene classification based on the existing bacterial taxonomy from ribosomal RNA databases, such as Ribosomal Database Project (RDP) (Cole et al. 2007), Greengenes (DeSantis et al. 2006a), and SILVA (Pruesse et al. 2007). Because the processing of taxonomy bins is rooted in polyphasic taxonomy (Colwell 1970), the taxonomy-supervised analysis is believed reflecting physiological, morphological, and genetic information (Sul et al. 2011). SortMeRNA (Kopylova et al. 2012) suited for closed-reference OTU clustering also belongs to the phylotyping methods that query sequences are searched against a reference database and then alignments are evaluated based on optimal regions of similarity between two sequences.
For the benefits and challenges of using phylotype-based methods, the interested reader is referred to the paper by Schloss and Westcott (2011) and references cited therein for more detailed description. For how to process taxonomic binning and overview of existing web tools for taxonomic assignment and phylotyping of metagenome sequence samples, the interested reader is referred to Dröge and McHardy (2012). For a comprehensive evaluation of the performance of open-source clustering software (namely, OTUCLUST, Swarm, SUMACLUST, and SortMeRNA) against UCLUST and USEARCH in QIIME, hierarchical clustering methods in mothur, and USEARCH’s most recent clustering algorithm, UPARSE, the interested reader is referred to Kopylova et al. (2016) and references cited therein. One example for comparison of various methods used to cluster 16S rRNA gene sequences into OTUs is from Jackson et al. (2016).
Within the framework of clustering-based OTU methods, various nonhierarchical clustering algorithms have been developed to improve the resolution of assigning 16S rRNA gene sequences to OTUs, including heuristic clustering, network-based clustering, and model-based clustering.
Although hierarchical clustering and heuristic clustering methods have been criticized due to either the computational complexity (hierarchical clustering) (Barriuso et al. 2011) or considerable loss in clustering quality (greedy clustering) (Sun et al. 2012; Chen et al. 2013a; Bonder et al. 2012), these two clustering methods are still used to assign OTUs in microbiome literature. In the following, we will introduce hierarchical clustering OTU methods (Sect. 8.1.2) and heuristic clustering OTU methods (Sect. 8.1.3), respectively.
8.1.2 Hierarchical Clustering OTU Methods
Single-linkage (also called nearest-neighbor) clustering, complete-linkage (also called furthest-neighbor) clustering, and average-linkage (also known as average-neighbor or UPGMA: unweighted pair-group method by using arithmetic averages) clustering are the three commonly used hierarchical clustering algorithms (Sokal and Rohlf 1995; Legendre and Legendre 1998). As we reviewed in Chap. 7, hierarchical clustering-based OTU methods are rooted in numerical taxonomy.
Single-linkage algorithm defines the distance between two clusters using the minimum distance between two sequences in each cluster. Complete-linkage algorithm defines the distance between two clusters using the maximum distance between two sequences in each cluster. Average-linkage algorithm defines the distance between two clusters using the average distance between each sequence in one cluster to every sequence in the other cluster. For more detailed description of these three clustering methods, the reader is referred to Chap. 7. Theses hierarchical clustering methods were widely adopted in the early development of microbiome software. For example, DOTUR (Distance-Based OTU and Richness) (2005) (Schloss and Handelsman 2005) is a clustering-based OTU method as the name suggested. It assigns sequences to OTUs by using either the furthest (i.e., complete-linkage), average (i.e., unweighted pair-group method by using arithmetic averages), or nearest (i.e., single-linkage) neighbor algorithm for each distance level (Schloss and Handelsman 2005). All these hierarchical clustering algorithms are commonly used in various disciplines (Sokal and Rohlf 1995; Legendre and Legendre 2012).
RDP (the Ribosomal Database Project) (2009) (Cole et al. 2009) is the taxonomy assignment alignment and analysis tools for quality-controlled bacterial and archaeal small-subunit rRNA research. RDP provides taxonomy-independent alignment, in which the trimmed reads are first aligned and then reads are clustered into OTUs at multiple pairwise distances using the complete-linkage clustering algorithm (Cole et al. 2009). In early release RDP has included a pairwise distance matrix suitable for the DOTUR molecular macroecology package (Cole et al. 2007). RDP pipeline was reviewed having better performance by using quality scores and performing a structural alignment but is only accurate at high sequence differences (Quince et al. 2009).
In 2009, in order to perform hypothesis tests and describe and compare communities using OTU-based methods, mothur (Schloss et al. 2009) integrated other software features into its implementation, including pyrosequencing pipeline (RDP) (Cole et al. 2009); NAST, SINA, and RDP aligners (Cole et al. 2009; DeSantis et al. 2006a, b; Pruesse et al. 2007); DNADIST (Felsenstein 1989); DOTUR (Schloss and Handelsman 2005); CD-HIT (Li and Godzik 2006); SONS (Schloss and Handelsman 2006b); LIBSHUFF (Schloss et al. 2004; Singleton et al. 2001); TreeClimber (Maddison and Slatkin 1991; Martin 2002; Schloss and Handelsman 2006a); and UniFrac (Lozupone and Knight 2005).
Although mothur includes many functions for performing hypothesis tests and describing and comparing communities, on the basis, mothur is the improved version of DOTUR: it uses the OTU-based methods of DOTUR (Schloss and Handelsman 2005) and CD-HIT (Li and Godzik 2006) for assigning sequences to OTUs, via implementing three de novo clustering algorithms, i.e., clustering sequences using the furthest, nearest, or UPGMA algorithms from DOTUR or using a nearest-neighbor-based approach from CD-HIT. Thus, mothur is one of representative hierarchical clustering methods for picking OTUs.
In 2009, ESPRIT (Sun et al. 2009) was proposed to use the k-mer (substrings of length k) distance to quickly compute pairwise distances of reads and classify sequence reads into OTUs at different dissimilarity levels via the complete-linkage algorithm. ESPRIT uses massively parallel pyrosequencing technology, i.e., via large collections of 16S rRNA pyrosequences for estimating species richness.
In 2010, SLP (a single-linkage pre-clustering) (Huse et al. 2010) was proposed to overcome the effect of sequencing errors and decrease the inflation of OTUs because the complete-linkage algorithm is sensitive to sequencing artifacts (Huse et al. 2010). SLP uses an average-linkage clustering to more accurately predict expected OTU richness in environmental samples.
In 2013, a modified version of mcClust (Cole et al. 2014; Fish et al. 2013) was proposed to allow the complete-linkage clustering to compute cluster and incorporate algorithmic changes for lowering the time complexity and speeding up clustering. HPC-CLUST (Matias Rodrigues and von Mering 2013) was proposed for large sets of nucleotide sequences via a distributed implementation of the complete- and average-linkage hierarchical clustering algorithms with high optimization.
In 2015, oclust (Franzén et al. 2015) was developed to improve OTU quality and decrease variance using long-read 16S rRNA gene amplicon sequencing by implementing the complete-linkage clustering.
8.1.3 Heuristic Clustering OTU Methods
In the early microbiome research, the commonly used hierarchical clustering methods have been adopted from numerical taxonomy (Sokal and Sneath 1963) and numerical ecology (Legendre and Legendre 1998). However, hierarchical clustering algorithms have the high computational complexity in time and space. Thus, to trade off between computational efficiency and accuracy, various heuristic methods have been proposed to reduce the computational complexity in sequence comparison by using greedy clustering strategy (e.g., CD-HIT (Huang et al. 2010; Li and Godzik 2006; Fu et al. 2012) and UCLUST (Edgar 2010)) instead of hierarchical clustering.
Heuristic clustering methods process input sequences one by one rather than compute pairwise distances of all sequences. Typically a greedy incremental clustering strategy and a seed are used to initiate its clustering (Chen et al. 2016). Because heuristic clustering methods just compare each sequence with the seed sequences, they are able to process massive sequence datasets. Thus, the heuristic clustering methods are considered as more attractive than clustering methods for picking OTUs in 16S rRNA datasets (Cai and Sun 2011; Wei et al. 2021) and have been applied by the human microbiome project (HMP) (Peterson et al. 2009).
Heuristic clustering methods for constructing OTUs
Heuristic clustering | References |
|---|---|
CD-HIT Aims to improve better accuracy, scalability, flexibility, and speed for clustering and comparing biological sequences Sorts the sequence before clustering by the length of sequences | |
USEARCH Is an algorithm for sequence database search that seeks high-scoring global alignments Aims to implement greedy clustering and typically achieves good sensitivity for identity Sorts the sequence before clustering by sequence abundance UCLUST Is a heuristic clustering algorithm that employs USEARCH to assign sequences to clusters Aims to take the advantages of UBLAST and USEARCH algorithms enabling sensitive local and global search of large sequence databases at exceptionally high speeds to achieve high throughput Both QIIME and UPARSE are based on USEARCH | Edgar (2010) |
GramClust Aims to use the inherent grammar distance metric of each pairwise sequence to cluster a set of sequences Determines partitioning for a set of biological sequences with higher statistical accuracy than both CD-HIT-EST and UCLUST | Russell et al. (2010) |
ESPRIT-Tree Is a new version of ESPRIT with focusing on the quasilinear time and space complexity Aims to avoid using the seed sequences to represent clusters like the most existing clustering methods Initially constructs a pseudometric-based partition tree for a coarse representation of the entire sequences Then iteratively finds the closest pairs of sequences or clusters and merges them into a new cluster Is able to analyze very large 16S rRNA pyrosequences in quasilinear computational time with the effectiveness and accuracy comparable to other greedy heuristic clustering algorithms | Cai and Sun (2011) |
DNACLUST Aims specifically to cluster highly similar DNA sequences for phylogenetic marker genes (e.g., 16S rRNA) Uses a greedy clustering strategy but via a novel sequence alignment and k-mer-based filtering algorithms to accelerate the clustering | DNAclust, Ghodsi et al. (2011) |
DySC (Dynamic Seed-based Clustering) Aims to use a dynamic seeding strategy for greedy clustering 16S rRNA reads First uses the traditional greedy incremental clustering strategy to form the pending clusters Then converts the pending cluster into a fixed cluster when it reaches a threshold size and reselects a new fixed seed The new fixed seed is defined as the sequence that maximizes the sum of k-mers shared between the fixed read and other reads in one cluster | Zheng et al. (2012) |
LST-HIT Is a DNA sequence clustering method based on CD-HIT Aims to speed up the computation due to the intrinsic difficulty in parallelization Uses a novel filtering technique based on the longest common subsequence (LST) to remove dissimilar sequence pairs before performing pairwise sequence alignment | Namiki et al. (2013) |
DBC454 Is a taxonomy-independent (i.e., unsupervised clustering) method for fungal ITS1 (internal transcribed spacer 1) sequences of 454 reads using a density-based hierarchical clustering procedure Aims to focus on reproducibility and robustness | Pagni et al. (2013) |
MSClust Is a multiseed-based heuristic clustering method Aims to use an adaptive strategy to generate multiseeds for one cluster Either assigns one query sequence to one cluster if the average distance between the sequence and seeds is smaller than the user-defined threshold Or marks the sequence as unassigned | Chen et al. (2013a) |
UPARSE Is an improved version of USEARCH with adding the chimera detection for seed sequences Aims to highly accurately sequence microbial amplicon reads and to reduce amplification artifacts Uses a greedy algorithm to perform chimera filtering and OTU clustering simultaneously through quality filtering, trimming reads, optionally discarding singleton reads, and then clustering the remaining reads Uses the similar algorithms that used in AmpliconNoise (Quince et al. 2011), i.e., inferring errors in a sequence using parsimony The generated OTU’s sequences were reported with ≤1% incorrect bases; such accurate OTUs are more closer to the species in a community | Edgar (2013) |
LSH Is a greedy clustering algorithm that uses the locality-sensitive hashing (LSH-based similarity function) to cluster similar sequences and make individual groups (OTUs) Aims to accelerate the pairwise sequence comparisons by using LSH algorithm and to improve the quality of sequence comparisons via incorporating a matching criterion The assigned OTUs can be utilized for computation of different species diversity/richness metrics (e.g., species richness estimation) | Rasheed et al. (2013) |
SUMACLUST Is a de novo clustering method Aims to perform exact sequence alignment, comparison, and clustering rather than semiglobal alignments implemented in CD-HIT and USEARCH Incrementally constructs the clusters via comparing an abundance-ordered list of input sequences with the representative already-chosen sequences based on a greedy clustering strategy SUMACLUST has been integrated into QIIME 1.9.0 | Mercier et al. (2013) |
Swarm Is a de novo clustering method based on an unsupervised single-linkage clustering method Aims to address two fundamental problems suffered by greedy clustering methods: arbitrary global clustering thresholds and input-order dependency induced by centroid selection First uses a local threshold clustering to generate an initial set of OTUs (nearly identical amplicons) by iteratively agglomerating similar sequences Then uses internal structure of clusters and amplicon abundances to refine the clustering results into sub-OTUs Swarm has been integrated into QIIME 1.9.0 | |
OTUCLUST Is a greedy clustering method (like SUMACLUST) Is a de novo clustering algorithm specifically designed for the inference of OTUs Aims to perform exact sequence alignment Compares an abundance-ordered list of input sequences with the representative already-chosen sequences Performs sequence de-duplication and chimera removal via UCHIME (Edgar et al. 2011) Is one component of software pipeline MICCA for the processing of amplicon metagenomic datasets that combines quality filtering, clustering of OTUs, taxonomy assignment, and phylogenetic tree inference | Albanese et al. (2015) |
VSEARCH Is an alternative to the USEARCH tool based on a heuristic algorithm for searching nucleotide sequences Aims to perform optimal global sequence alignment of the query using full dynamic programming Performs searching, clustering, chimera detection (reference-based or de novo) and subsampling, paired-end reads merging, and dereplication (full length or prefix) | VSEARCH (Rognes et al. 2016) |
OptiClust Is an OTU assignment algorithm that iteratively reassigns sequences to new OTUs to maximize the Matthews correlation coefficient (MCC) Aims to improve the quality of the OTU assignments Requires a distance matrix for constructing OTUs because it is a distance-based algorithm | Westcott and Schloss (2017) |
ESPRIT-Forest Is an improved method of ESPRIT and ESPRIT-Tree Aims to cluster massive sequence data in a sub-quadratic time and space complexity while inheriting the same pipeline of ESPRIT and ESPRIT-Tree for preprocessing, hierarchical clustering, and statistical analysis First organizes sequences into a pseudometric-based partitioning tree for sublinear time searching of nearest neighbors Then uses a new multiple-pair merging criterion to construct clusters in parallel using multiple threads | Cai et al. (2017) |
DBH Is a de Bruijn (DB) graph-based heuristic clustering method Aims to reduce the sensitivity of seeds to sequencing errors by just selecting one sequence as the seed for each cluster First forms temporary clusters using the traditional greedy clustering approach Then builds a DB graph for this cluster and generates a new seed to represent this cluster when the size of a temporary cluster reaches the predefined minimum sequence number | Wei and Zhang (2017) |
Fuzzy Aims to improve the clustering quality via the clustering based on fuzzy sets Takes into account uncertainty when producing OTUs using the fuzzy OTU-picking algorithm | Bazin et al. (2019) |
DMSC Is a dynamic multiseed clustering (DMSC) method for generating OTUs First uses greedy incremental strategy to generate a series of clusters based on the distance threshold When the sequence number in a cluster reaches the predefined minimum size, then the multicore sequence (MCS) is selected as the seeds of the cluster The average distance to MCS and the distance standard deviation in MCS will determine whether a new sequence is added to the cluster The MCS will be dynamically updated until no sequence is merged into the cluster | Wei and Zhang (2019) |
8.1.4 Limitations of Clustering-Based OTU Methods
The early methodological developments of numerical taxonomy provided a rich source for modern ecology and microbiome research. For example, the concepts of OTU, similarity, and using similarity to define taxonomic rank and using distance as a measure of taxonomic similarity have been adopted in current ecology and microbiome research.
However, OTU methods in microbiome and numerical taxonomy have different assumptions, purposes, and approaches. Numerical taxonomy assumes that characters can be grouped based on their similarities. It employs commonly used clustering to perform classification of characters. Bioinformatic analysis of microbiome assumes that sequence similarity can predict taxonomic similarity. It has dual purpose: grouping sequences and estimating diversity. Its final goal is to detect the association between microbiome health and the development of the disease. Thus, microbiome research is not restricted to commonly used clustering and heuristic clustering.
Theoretically any methods that can be used to achieve the goals are appropriate. The newly developed methods such as denoising, refining OTU clustering, model-based methods, and filtering-free methods are all targeting the final goal of bioinformatics in microbiome research.
However, the main motivation of moving beyond the clustering-based OTU methods in microbiome research is that current application of OTU methods to microbiome data has several limitations. Here we describe and discuss the clustering-based OTU methods and especially their limitations that have motivated microbiome research to move beyond them.
8.1.4.1 Assumptions of OTU Methods in Numerical Taxonomy and Microbiome
OTU methods used in numerical taxonomy and microbiome have different aims. Adopting OTU methods into microbiome studies is somewhat inappropriate and thus is controversial.
Although at the beginning the concept of OTU is convenient for bioinformatic analysis of microbiome sequencing data, the use of the OTU methods in microbiome literature deviates from the considerations of the original authors. As reviewed in Chap. 7, the original authors of numerical taxonomy strictly separated taxonomic classifications from phylogenetic classifications and treated the latter as “phylogenetic speculation” and the former as “taxonomic procedure.” The original authors considered OTUs as operational not functional; the functionality remained to taxa; cluster analysis of similarity is merely on datasets at hand and requires that the characters are from larger and randomly chosen samples; taxonomic similarity does not represent the phylogenetic similarity. In contrast, by assigning sequences to a particular OTU, actually it assumes that the similarities among sequences are closely related to the phylogeny, and therefore the similarities of sequences can most likely be used to derive phylogenetic divergence or ecologically rank the similarity of taxa. However, the assumption that sequence similarity of 16S hypervariable regions is a good proxy for phylogenetic and therefore ecological similarity is problematic (Prosser et al. 2007; Tikhonov et al. 2015; Preheim et al. 2013) because sequence similarity does not imply ecological similarity. It was demonstrated that sequence similarity very poorly predicted ecological similarity (Tikhonov et al. 2015). The application of OTU methods in microbiome studies actually forces OTUs to play the functional role of taxa.
8.1.4.2 Challenges of Defining Species Using OTU Methods
OTU methods have been used in microbiome studies to classify taxa in classical Linnaean taxonomy at different taxonomic levels with similar DNA sequences.
Because 16S rRNA genes evolve at different rates in different organisms, determining taxonomic rank by sequence identity thresholds is approximate at the best (Woese 1987). Hierarchical clustering methods generally use a predefined clustering threshold to the hierarchical tree and then group sequences within the threshold into one OTU. For example, because a natural entity “species” cannot be identified as a group of strains that is genetically well separated from its phylogenetic neighbors, sequences are often clustered into OTUs as proxies for species, although sequence divergence is not evenly distributed in the 16S rRNA region. Thus, defining a species by a polyphasic approach is only a pragmatic approach.
Previously, the threshold value of 70% was used in the phylogenetic definition of a species (“approximately 70% or greater DNA-DNA relatedness and with 5°C or less”) (Wayne et al. 1987). This approach of applying DNA similarity to study bacterial species was well received and well proven and has been acknowledged since the 1970s (Stackebrandt and Goebel 1994; Johnson 1985; Steigerwalt et al. 1976). In 1994 Stackebrandt and Goebel (1994) proposed to use the “similarities of 97% and higher” sequence identity as the canonical clustering threshold when few 16S rRNA sequences were available. In practice the 3% dissimilarity is often chosen as the cutoff value to define bacteria species (Sogin et al. 2006; Sun et al. 2009; Schloss 2010; Huse et al. 2007) although different cutoff values of similarity have been studied such as 99.6–95.6% similarity (equating 1–13 bp) (Hathaway et al. 2017).
However, the cutoff value of similarities of 97% was based on the observation that species having 70% or greater DNA similarity usually have more than 97% sequence identity with these 3% or 45-nucleotide differences being concentrated mainly in certain hypervariable regions (Stackebrandt and Goebel 1994) given the primary structure of the 16S rRNA is highly conserved. Thus, the criterion of 97% similarities or 3% dissimilarities (Schloss and Handelsman 2005) is observed and experienced. It is not really able to define a natural entity “species.” Especially, it was shown that 97% threshold is not optimal for an approximation to species because it is too low. To define a species, at least 99% of OTUs should be used (Edgar 2018). In summary, this practical approach of using OTUs to define a species is some kinds of arbitrary, subjective, and therefore inaccurate.
8.1.4.3 Criteria of Defining the Levels of Taxonomy
Like the 3% dissimilarities of sequences that are used to define a species (Borneman and Triplett 1997; Stackebrandt and Goebel 1994; Hugenholtz et al. 1998; Konstantinidis et al. 2006; Goris et al. 2007), accordingly in practice the 95% sequence similarity (95% homology) or the dissimilarities of 5% by species are used to define a genus (Wolfgang Ludwig et al. 1998; Everett et al. 1999; Sait et al. 2002; Schloss and Handelsman 2005); 10% dissimilarities (90% homology) are used to define a family (Schloss et al. 2009); 20% dissimilarities (80% homology) are used to define a phylum (Schloss and Handelsman 2005); an interkingdom identity range of 70–85% is used to define a kingdom (Woese 1987; Borneman and Triplett 1997); and 45% dissimilarities (55% homology) are used to define a domain (Stackebrandt and Goebel 1994).
This practice of using an arbitrary fixed global clustering threshold to group amplicons into molecular OTUs has been criticized (Mahé et al. 2015b) because global clustering thresholds have rarely been justified and are not applicable to all taxa and marker lengths (Caron et al. 2009; Nebel et al. 2011; Dunthorn et al. 2012; Brown et al. 2015). Actually it is impossible to use an accurate distance-based threshold to define taxonomic levels (Schloss and Westcott 2011). Therefore, the criteria of defining the levels of taxonomy are arbitrary, subjective, and inaccurate.
8.1.4.4 Clustering Algorithms and the Accuracy of OTU Clustering Results
As reviewed in Chap. 7, the original OTU methods are based on cluster analysis. However, when the clustering-based OTU methods were adopted in microbiome studies, several limitations have been detected that are associated with the clustering algorithms.
In practice it is difficult to choose which clustering algorithms to use even if we could use a constant threshold to define OTUs. Because, in general, different clustering algorithms have assessed having different clustering quality, such as by comparing hierarchical and heuristic clustering algorithms in OTU construction, hierarchical clustering algorithms are assessed as more accurate (Sun et al. 2010). However, heuristic clustering methods are able to process massive sequence datasets. Thus, heuristic clustering methods are more attractive than hierarchical clustering methods for picking OTUs in 16S rRNA datasets (Cai and Sun 2011; Wei et al. 2021) and have been applied by the human microbiome project (HMP) (Peterson et al. 2009). However, most heuristic clustering methods just randomly select one sequence as the seed to represent the cluster and use this seed fixedly which results in the assigned OTUs sensitive to/depending on the selected seeds. Actually no real consensus has achieved on how the random seed is chosen, suggesting that OTU assignments are not definitive (Schloss and Westcott 2011).
Among the commonly used hierarchical clustering algorithms, a unanimous conclusion on which hierarchical clustering algorithm is optimal has not been reached.
The complete-linkage was originally recommended in numerical ecology because it is often desirable in ecology when one wishes to delineate clusters with clear discontinuities (Legendre and Legendre 1998), while the single-linkage clustering is implemented (Wei et al. 2012) in BlastClust (BLASTLab 2004) and GeneRage (Enright and Ouzounis 2000). Other researchers demonstrated that the average-linkage algorithm produces more robust OTUs than other hierarchical and heuristic clustering algorithms (i.e., CD-HIT, UCLUST, ESPRIT, and BlastClust) (Schloss and Westcott 2011; Sun et al. 2012; Quince et al. 2009). The average-linkage algorithm was further evaluated as having higher clustering quality compared to the other common clustering algorithms and even compared to greedy heuristic clustering algorithms, including distance-based greedy clustering (DGC) and abundance-based greedy clustering (AGC) (Westcott and Schloss 2015; Ye 2011). In such analysis, an OTU is generally considered presenting a bacterial species (Kuczynski et al. 2012). Therefore, using the average-linkage algorithm for clustering 16S rRNA gene sequences into OTUs has been suggested (Huse et al. 2010; Schloss and Westcott 2011). The suggestion of using the average-linkage clustering in microbiome data is in line with the suggestion of the original authors of numerical taxonomy (see Chap. 7 for details).
The accuracy of clustered OTUs is also determined by removal of low-quality reads and of chimera sequences. It was shown that strict quality filtering may lead to a loss of information, which particularly affects the least abundant species, whereas if reads are clustered with low quality, then spurious OTUs may be produced which may result in overestimating the complexity of the analyzed samples (Albanese et al. 2015). It was also shown that amplicon input order can result in controversial results of OTU assignments (Koeppel and Wu 2013; Mahé et al. 2014, 2015b).
Different clustering algorithms and associated clustering thresholds have significantly impacted on the performance of clustering OTUs (Franzén et al. 2015). Since actually optimal thresholds are all higher than 97% (Edgar 2018). Thus, algorithms cannot be meaningfully ranked by OTU quality at 97% of similarity. For example, when comparing clustering algorithms, single-linkage, complete-linkage, average-linkage, AGC (Ye 2011), and OptiClust (OC) (Westcott and Schloss 2017), it was shown that no algorithm is consistently better than any other (Edgar 2018). In summary, no real consensus has emerged on the clustering algorithm.
The controversial and challenges of clustering approaches are due to using different criteria to assess clustering quality or test accuracy of clustering, such as simulation, information theoretic-based normalized distance measurement (Chen et al. 2013b; Baldi et al. 2000; van Rijsbergen 1979), stability of OTU assignments (He et al. 2015), and Matthew’s correlation coefficient (MCC) (Schloss and Westcott 2011; Westcott and Schloss 2015), or combining the MCC and the F-score (Preheim et al. 2013; Baldi et al. 2000).
Different criteria often reach totally different conclusions. For example, many hierarchical and greedy clustering methods are unable to stably assign or produce OTUs. Thus, some researchers, such as He et al. (2015), thought that clustering all sequences into one OTU is completely useless for downstream analysis. By using MCC, Westcott and Schloss (2015) further demonstrated that the stability of OTUs actually suffered a lack of quality because the stability concept focused on the precision of the assignments and did not reflect the quality of the OTU assignments. Actually, this point was also raised in the numerical taxonomy.
In summary, the commonly used hierarchical and heuristic-based OTU methods suffered several fundamental problems, such as (1) the assumption of sequence similarity implying ecological similarity, (2) using arbitrary criteria to define the level of taxonomy (e.g., the dissimilarity of 3% OTUs is used to define species), (3) seed sensitivity, (4) input-order dependency, and (5) read preprocessing.
8.1.5 Purposes of Using OTUs in Microbiome Study
In microbiome study, the concept of OTUs was borrowed from numerical taxonomy. OTUs are used to serve the triple purpose: First is to define different taxonomic levels of taxonomy. Second is expected to reduce the impact of amplicon sequencing error on ecological diversity estimations and measures of community composition via grouping errors together with the error-free sequence (Eren et al. 2016). Third, the refined OTUs were repurposed to play the functionality of species.
8.1.5.1 Defining Taxonomy
Initially when the OTU concept was proposed to be used in microbiome sequencing, the purpose is for the taxonomic grouping, i.e., identifying bacterial population boundaries (Callahan et al. 2017). However, as we reviewed above and others such as Murat Eren et al. (2016) and Benjamin Callahan et al. (2017), the original purpose of using OTUs has not been achieved by the clustering OTU methods. For example, it is problematic to define OTUs using 97% sequence similarity threshold in the closed-reference approach because two sequences might be 97% similar to the same reference sequence, maybe less similarity (e.g., only 94%) shared by each other (de Queiroz and Good 1997; Westcott and Schloss 2015). Thus, the use of 97% OTUs to construct “species” has not been largely approved. In practice a 16S rRNA gene sequence similarity threshold range of 98.7–99% was recommended for testing the genomic uniqueness of a novel isolate(s) (Erko Stackebrandt and Ebers 2006). Similarly, it has not been largely approved to use the 95%, 90% of sequence similarity to define a genus and a family, respectively, and so on.
8.1.5.2 Reducing the Impact of Sequencing Error
The second purpose of using OTU concept and methods is to differentiate true diversity from sequencing errors (Callahan et al. 2017). This purpose is different from and orthogonal to the first one. When the OTU concept was initially proposed and used in microbiome study, OTUs were not expected to reduce the impact of amplicon sequencing error. The task of correcting errors has been repurposed to OTU (Rosen et al. 2012). In other words, this task is an expectation or was weighted as having the function of reducing sequence errors because the use of 97% sequence similarity threshold has successfully reduced the impact of erroneous OTUs on diversity estimations (Eren et al. 2016). After the second role was repurposed to OTUs, clustering reads into OTUs by sequence similarity became a standard approach to filter the noise (Tikhonov et al. 2015).
However, the second purpose has not be achieved either. The use of OTUs for differentiating true diversity from sequencing errors is only appropriate for higher levels of taxonomy (e.g., phylum). When OTU methods were used for probing finer-scale diversity in lower levels of taxonomy, they have intrinsically high false-positive and false-negative rates because using arbitrary cutoff values not only overestimate diversity when there exist errors larger than the OTU-defining cutoff but also cannot resolve real diversity at a scale finer (Rosen et al. 2012). It was also shown that clustering reads into OTUs greatly underestimates ecological richness (Tikhonov et al. 2015). In other words, the use of OTUs has not reduced the controversy on diversity estimations and measures of community composition.
Taken together, both purposes of using OTU-based methods have problems to be achieved and the two major challenges remained: one is to identify bacterial population boundaries and another is to differentiate true diversity from sequencing errors (Preheim et al. 2013).
The third purpose of using OTUs to play the functionality of species in microbiome research is more complicated. To better understand whether this purpose is achieved, we write Sect. 8.1.6 to provide a theoretical background to discuss definitions of species and species-level analysis.
8.1.6 Defining Species and Species-Level Analysis
Defining species is a core component of defining taxonomy, which is an important purpose that the 16S rRNA approach assumed to achieve. In Chap. 7, we reviewed three major species concepts that have been suggested to distinguish species in taxonomic studies: (1) genospecies (a group of organisms exchanging genes), (2) taxospecies (a phenetic cluster), and (3) nomenspecies (organisms bearing the same binomen). We also discussed the difference between a phenetic species and a biological species.
Historically the prokaryote taxonomy was reviewed (Rosselló-Mora and Amann 2001) having largely focused on the nomenclature of taxa (Heise and Starr 1968; Buchanan 1955; Cowan 1965; Sneath 2015; Trüper 1999) rather than the practical circumscription of the species concept applied to prokaryotes. Today’s prokaryotic species concept results from empirical improvements of what has been thought to be a unit (Rosselló-Mora and Amann 2001). However, the underlying idea of defining a species is not only to formulate a unique unit for analysis but also to determine the functionality/genomic (i.e., a genotypic/genomic cluster definition) and phenotypic properties (i.e., not only biochemical or physiological properties but also chemo-taxonomical markers, such as fatty acid profiles that are related to the species and its host).
The topics on eukaryotic and prokaryotic species concepts are very complicated and still debatable. Many books and review articles have contributed to these topics. We here have no intention of fully reviewing either eukaryote or prokaryotic species concept. The interested reader is referred to these books (Claridge et al. 1997b; Wheeler and Meier 2000) and articles (Rosselló-Mora and Amann 2001; Sokal and Crovello 1970; Mishler 2000; Hey 2006; Zink and McKitrick 1995; Balakrishnan 2005; Platnick 2000; Mishler 1999). Below we just summarize various definitions of species and provide a history background of species concepts in microbiome study.
8.1.6.1 Eukaryote Species Concepts
The species concept has been discussed among microbiologists and eukaryote taxonomists (Claridge et al. 1997a). The species concepts for prokaryotes and eukaryotes are different (May 1986, 1988); briefly review of species concept for eukaryotes will definitely help to delineate the species concept for prokaryotes.
Selective eukaryote species concepts
Biological species concept (BSC) |
BSC defines a species as “groups of actually or potentially interbreeding natural populations which are reproductively isolated from other such groups” (Mayr 1940, 1942) and (Mayr 1963) (p. 19) In his later publications (Mayr 1969, 2004, 2015), Mayr dropped the much criticized phrase “potentially interbreeding” from the definition and defined a species as: “groups of interbreeding natural populations that are reproductively (genetically) isolated from other such groups” (Mayr 2004) |
Evolutionary species concept (ESC) |
ESC defines a species as “a lineage (an ancestral-descendant sequence of populations) evolving separately from others and with its own unitary evolutionary role and tendencies” (Simpson 1961) “a single lineage of ancestor-descendant populations which maintains its identity from other such lineages and which has its own evolutionary tendencies and historical fate” (Wiley 1978) “an entity composed of organisms which maintains its identity from other such entities through time and over space, and which has its own independent evolutionary fate and historical tendencies” (Mayden 1997; Wiley and Mayden 2000) |
Phenetic or polythetic species concept (PhSC) |
PhSC defines a species as “the species level is that at which distinct phenetic clusters can be observed” (Sneath 1976) (p. 437) |
Phylogenetic species concept (PSC) |
PSC defines a species as “the smallest biological entities that are diagnosable and/or monophyletic” (Mayden 1997) “the smallest diagnosable monophyletic unit with a parenteral pattern of ancestry and descent” (Rosselló-Mora and Amann 2001) |
8.1.6.2 Prokaryote or Bacterial Species Concepts
- 1.
The 1870s to Early 1950s: Phenotypic Classification (Phenotypic Definition of Species)
The ability to isolate organisms in pure cultures started in 1872 cultivating pure colonies of chromogenic bacteria by Joseph Schroeter (Logan 2009). Cultivating microorganisms facilitates bacteria classification based on the phenotypic description of these organisms. However, early bacterial species was often defined based on monothetic groups (a unique set of features). This is a phenotypic definition of species, which lies on a database with an accurate morphologic and phenotypic (e.g., biochemical) description of type strains or typical strains and comparing the isolate to be identified to the database using standard methods to determine these characteristics for the isolate (Clarridge 2004). The limitation is that these sets of phenotypic properties were subjectively selected (Goodfellow et al. 1997).
Among the more than 20 species concepts, Mayr’s BSC is the most widely known and most controversial concept (Rosselló-Mora and Amann 2001; Sokal 1973). BSC attempts to unify genetics, systematics, and evolutionary biology (Claridge et al. 1997a). However, it lacks of practicability and hence generally it was agreed that Mayr’s BSC should be abandoned (Rosselló-Mora and Amann 2001; Sokal 1973). Therefore, BSC cannot presently be applied to prokaryotes (Stackebrandt and Goebel 1994).
- 2.
The Late 1950s to Early 1960s: Phenetic Classification
The development of numerical taxonomy in the late 1950s in parallel to application of computer for multivariate analyses makes bacteria classification toward an objective approach (Sneath 1989). The numerical taxonomy began to establish when modern biochemical analytical techniques advanced to study the distributions of specific chemical constituents (i.e., amino acids, proteins, sugars, and lipids in bacteria) (Rosselló-Mora and Amann 2001; Logan 2009).
- 3.
The Late 1960s: Genomic Classification
- 4.
The Late 1970s to the Mid-1980s: 16S rRNA Classification
In the late 1970s, with the advent of cataloging ribosomal ribonucleic acids (rRNAs) (Stackebrandt et al. 1985), the rRNA and especially 16S rRNA sequences were shown to be a very useful molecular marker for phylogenetic analyses (Ludwig and Schleifer 1994).
Clustering-based OTU methods have been combined with the 16S rRNA sequencing. The 16S rRNA sequencing method has been widely used for the prokaryotic classification (Olsen et al. 1994), and the datasets generated from 16S rRNA sequencing studies have also been increasingly used to propose new bacterial species (Stackebrandt and Goebel 1994).
The determination of relationships between distantly related bacteria has been evidenced a remarkable breakthrough in the late 1970s by cataloging 16S ribosomal ribonucleic acids (rRNAs) (Stackebrandt et al. 1985) and DNA-RNA hybridization (De Ley and De Smedt 1975) and in the mid-1980s by the full sequence analysis of rRNA (Rosselló-Mora and Amann 2001). In the 1980s, determining phylogenetic relationships of bacteria and all life forms by comparing a stable part of the genetic code (commonly the 16S rRNA gene) became a new standard for identifying bacteria (Woese et al. 1985; Woese 1987).
- 1.
It is highly conserved (Dubnau et al. 1965; Woese 1987). Given its conservation the 16S rRNA gene marks evolutionary distance and relatedness of organisms; thus the evolutionary rates in the 16S rRNA gene sequence can be estimated through comparing studies of nucleotide sequences (Kimura 1980; Pace 1997; Thorne et al. 1998; Harmsen 2004). Currently it has accepted that 16S rRNA sequence analysis can be used for prokaryotic classification to identify bacteria or to assign close relationships at the genus and species levels (Clarridge 2004).
- 2.
It is universal in bacteria; thus the relationships among all bacteria can be measured (Woese et al. 1985; Woese 1987). This is also an important property. It was reviewed that through the comparison of the 16S rRNA gene sequences, it not only can classify strains at multiple levels, including the species and subspecies level, but also differentiate organisms at the genus level across all major phyla of bacteria (Clarridge 2004).
Due to above two important properties, bacterial phylogeny (the genealogical trees among the prokaryotes) can be established by comparing the 16S rRNA gene sequences (Ludwig et al. 1998). PSC includes monophyletic species concept (MSC) and diagnostic species concept (DSC) (Hull 1997). Both are defined as phylogenetic (or genealogical) concepts with a minimal time dimension (Rosselló-Mora and Amann 2001).
When DNA was discovered, prokaryote classification was based solely on phenotypic characteristics. With the development of numerical taxonomy (Sneath and Sokal 1973), the individuals are treated as operational taxonomic units that are polythetic (they can be defined only in terms of statistically covarying characteristics), resulting in a more objective circumscription of prokaryotic units. The discovery of genetic information gave a new dimension to the species concept for microorganisms, which enable at least a first rough insight into phylogenetic relationships. Thus, the species concept for prokaryotes evolved into a mostly phenetic or polythetic. This means that species are defined by a combination of independent, covarying characters, each of which may occur also outside the given class, thus not being exclusive of the class (Van Regenmortel 1997).
Compared to DNA-RNA hybridization approach, the 16S rRNA gene sequence approach is much simpler and thus has replaced DNA-RNA hybridization and become the new gold standard to define a species (Fournier et al. 2003; Harmsen 2004). However, as we will describe in Sect. 8.1.6.3, using the 16S rRNA method to define a species still remains challenging. Before we move forward to definition of species in 16S rRNA method, we quote some microbiological definitions of species.
“a microbial species is a concept represented by a group of strains, that contains freshly isolated strains, stock strains maintained in vitro for varying periods of time, and their variants (strains not identical with their parents in all characteristics), which have in common a set or pattern of correlating stable properties that separates the group from other groups of strains (Gordon 1978).”
“a group of strains that show a high degree of overall similarity and differ considerably from related strain groups with respect to many independent characteristics (Colwell et al. 1995)” or
“a collection of strains showing a high degree of overall similarity, compared to other, related groups of strains (Colwell et al. 1995).”
8.1.6.3 16S rRNA Method and Definition of Species
Using the 16S rRNA similarities to define bacteria species remains challenging. Much early in the 1870s, Ferdinand Cohn investigated whether the similarity exits between bacteria and whether bacteria, like animals and plants, can be arranged in distinct taxa (Cohn 1972; Schlegel and Köhler 1999). Cohn considered the form genera as natural entities but considered species as largely artificial. Cohn emphasized that character of the classification system is artificial. The original authors of OTU method Sokal and Sneath (1963) also criticized their then current taxonomy made little increase in understanding the nature and evolution of the higher categories (Sokal and Sneath 1963) (p. 5).
When clustering-based OTU methods emerged into the 16S rRNA sequencing analysis, an important concept, the phylogenetic species concept (PSC), was developed. PSC compares the 16S rRNA similarities with DNA similarity (DNA-DNA similarities), assuming that nucleic acid (DNA) reassociation can determine whether taxa are phylogenetically homogeneous. PSC defines a species that would generally include strains with “approximately 70% or greater DNA-DNA relatedness and with 5°C or less ΔTm” (Wayne et al. 1987). Here, Tm is the melting temperature or the thermal denaturation midpoint, and ΔTm is the difference between the homoduplex DNA Tm and the heteroduplex DNA Tm. Thus, ΔTm is a reflection of the thermal stability of the DNA duplexes.
However, as reviewed in Sect. 8.1.4.2, the rationale for using DNA reassociation as the gold standard to delineate species originates from the observations that a high degree of correlation existed between DNA similarity and chemotaxonomic, genomic, serological, and numerical phenetic similarity (Stackebrandt and Goebel 1994). The 16S rRNA sequencing method does not have sufficient power to correctly define bacterial species, which was reviewed in several articles including Ash et al. (1991); Amann et al. (1992); Fox et al. (1992); Martinez-Murcia et al. (1992); Stackebrandt and Goebel (1994); and Johnson et al. (2019).
- 1.
The 97% sequence similarity used by 16S rRNA method is based on the assumption that 70% DNA similarity defines a species. Organisms that have 70% or greater DNA similarity will not necessarily have at least 96% DNA sequence identity. The threshold value of 70% does not consider the possibility that the tempo and mode of changes differ in different prokaryotic strains (Stackebrandt and Goebel 1994). DNA hybridization is significantly higher resolution of power than that of sequence analysis (Amann et al. 1992). Thus, the 70% DNA similarity cannot be used to define a species. Moreover, species having 70% DNA similarity usually have more than 97% sequence identity (Stackebrandt and Goebel 1994). Thus, even if the 70% DNA similarity can be used to define a species, the 97% sequence identity cannot be used to define a species.
- 2.
The 16S rRNA has a highly conserved primary structure; there does not exist a linear correlation between the two phylogenetic parameters DNA-DNA similarity percent and 16S rRNA similarity for closely related organisms (Stackebrandt and Goebel 1994). Sequence similarities and DNA reassociation values obtained for the same strain pairs do not have a linear relationship (Erko Stackebrandt and Ebers 2006; Stackebrandt and Goebel 1994), and actually 16S rRNA sequence identity is not sufficient to guarantee species identity (Fox et al. 1992; Rosselló-Mora and Amann 2001; Martinez-Murcia et al. 1992).
- 3.
The 3% or 45-nucleotide differences are not evenly distributed over the primary structure of the molecule; instead they are highly likely to concentrate in the hypervariable regions. Thus, it is impossible to exactly describe the genealogy of the molecule because the false identities are simulated and the actual number of evolutionary events is masked (Grimont 1988; Stackebrandt and Goebel 1994).
- 4.
Technically, 16S variable regions (i.e., ranging from single variable regions, such as V4 or V6, to three variable regions, such as V1–V3 or V3–V5) cannot be targeted with short-read sequencing platforms (e.g., using Illumina sequencing platform to produce ≤300 bases of short sequences) to achieve the taxonomic resolution afforded by sequencing the entire (~1500 bp) gene (Johnson et al. 2019).
- 5.
Particularly, the examples described below (Rosselló-Mora and Amann 2001) suggest it is difficult to use the 16S rRNA similarities to define bacteria species, because (1) different species could have identical (Probst et al. 1998) or nearly identical 16S rRNA sequences (Fox et al. 1992; Martinez-Murcia et al. 1992); (2) within a single species, there exists a microheterogeneity of the 16S rRNA genes (Bennasar et al. 1996; Ibrahim et al. 1997); and (3) in exceptional cases, single organisms with two or more 16S rRNA genes could have relatively high sequence divergence (Mylvaganam and Dennis 1992; Nübel et al. 1996).
In summary, clustering sequence similarity of 16S rRNA can never be solely used to define bacterial species. On the one hand, it was recognized that the DNA hybridization (DNA reassociation) remains the optimal method for measuring the degree of relatedness between highly related organisms and hence cannot be replaced by 16S rRNA method for defining species (Stackebrandt and Goebel 1994; Rosselló-Mora and Amann 2001). On the other hand, it has been accepted that a prokaryotic species classification should be defined by integrating both phenotypic and genomic parameters, as well as should analyze and compare as many phenotypic and genomic parameters as possible (Wayne et al. 1987). This approach is known as “polyphasic taxonomy” (Vandamme et al. 1996). Based on this polyphasic approach, in 2001 a more pragmatic prokaryote species concept (called a phylo-phenetic species concept) has been described as “a monophyletic and genomically coherent cluster of individual organisms that show a high degree of overall similarity in many independent characteristics, and is diagnosable by a discriminative phenotypic property” (Rosselló-Mora and Amann 2001). Obviously, this is far beyond the clustering-based OTU method can achieve.
8.1.6.4 16S rRNA Method and Physiological Characteristics
Whether the 16s rRNA method can inference the physiological characteristics is debatable. On the one hand, the 16S rRNA approach has the practical advantage for species identification and has been used frequently in bacterial phylogenetics, species delineation, and microbiome studies; for example, it is recommended to include the ribosomal sequence for describing new prokaryotic species (Ludwig 1999; Chakravorty et al. 2007). Some researchers considered that comparative analysis of 16S rRNA is a very good method to find a first phylogenetic affiliation in both potentially novel and poorly classified organisms (Goodfellow et al. 1997). Thus, although the 16S rRNA sequence analysis is not sufficient for numerically delineating borders of the prokaryotic species, it can indicate the ancestry pattern of the taxon studied, as well as confirm the monophyletic nature in grouping the members (Rosselló-Mora and Amann 2001).
On the other hand, other researchers have questioned the validity of 16S rRNA as a marker for phylogenetic inferences and thought that it is very difficult to infer the physiological characteristics of an organism by clustering the rRNA sequence method (Rosselló-Mora and Amann 2001; Cohan 1994; Gupta 1998; Gribaldo et al. 1999). Thus, using arbitrary threshold of 97% similarity (or 3% dissimilarity) to define OTUs that could be interpreted as a proxy for bacterial species is computationally convenient, but at the expense of accurate ecological inference (Eren et al. 2016) because 3% OTUs are often phylogenetically mixed and inconsistent (Koeppel and Wu 2013; Nguyen et al. 2016; Eren et al. 2014).
Recently, Hassler et al. (2022) found that phylogenies of the 16S rRNA gene and its hypervariable regions lack concordance with core genome phylogenies, and they concluded that the 16S rRNA gene has a poor phylogenetic performance and has far-reaching consequences.
By comparing core gene phylogenies to phylogenies constructed using core gene concatenations to estimate the strength of signal for the 16S rRNA gene, its hypervariable regions, and all core genes at the intra- and inter-genus levels, Hassler et al. (2022) showed that at both intra- and inter-genus taxonomic levels, the 16S rRNA gene was recombinant and subject to horizontal gene transfer, suffering from intragenomic heterogeneity, accumulating recombination and an unreliable phylogenetic signal. The poor phylogenetic performance using the 16S rRNA gene not only results in incorrect species/strain delineation and phylogenetic inference but also has the potential to confound community diversity metrics if incorporating phylogenetic information in the analysis such as using Faith’s phylogenetic diversity and UniFrac (thus, their use to measure the diversity is not recommended) (Hassler et al. 2022). Additionally, taxonomic abundance and hence measures of microbiome diversity may also be inflated and confounded by the multiple copies within a genome and wide range in 16S rRNA gene among genomes (De la Cuesta-Zuluaga and Escobar 2016; Louca et al. 2018).
8.2 Moving Toward Single-Nucleotide Resolution-Based OTU Methods
In summary, using OTUs as the atomic unit of analysis has both benefits and controversies. This consequence is often due to unacknowledged factor that OTUs fail to serve the dual tasks well; and especially the connection between OTUs and species is largely unfounded (Stackebrandt and Ebers 2006). In order to overcome the limitations of clustering-based OTU methods in bioinformatic analysis of microbiome sequencing data, the reform movements of moving beyond OTUs and clustering-based OTU methods have begun in microbiome research. Two strategies for these reform movements that represent overlapping goals are moving toward single-nucleotide resolution and moving beyond the OTU-based methods with denoising-based methods. In this section, we describe the first strategy, moving toward single-nucleotide resolution-based OTU methods, and in Sect. 8.3, we investigate the second strategy: moving beyond the OTU-based methods with denoising-based methods.
8.2.1 Concept Shifting in Bioinformatic Analysis
In bioinformatic analysis of microbiome data, a concept shifting regarding analysis units began with a dissatisfactory of using similarity/dissimilarity threshold to define OTUs.
As we reviewed in Sects. 8.1.5 and 8.1.6, OTUs used in microbiome study have three purposes: (1) to define taxonomic levels, (2) to improve the accuracy of ecological diversity estimations and measures of community composition, and (3) to play the functionality of species. All these are done through defining certain particular taxonomic levels by borrowing ecological taxonomic concepts into the context of high-throughput marker-gene sequencing of microbial communities, such as to classify microbial species (Rosen et al. 2012); the 97% sequence identity (or 3% ribosomal) OTUs are used to define the level of “like species.” When we do this way, we actually assume that the greater similar sequences more likely represent phylogenetically similar organisms. Defining OTUs in this way is expected to simplify the complexity of the large datasets (Westcott and Schloss 2015), to facilitate taxonomy-independent analyses, such as alpha diversity, beta diversity, and taxonomic composition, and thus to effectively reduce the computational resources (Human Microbiome Project 2012; He et al. 2015).
However, it was assessed (Chen et al. 2013b) that using a constant threshold to define OTUs at a specific taxonomic level, e.g., using 3% dissimilarity to define species, is not ideal because clustering reads into OTUs vastly underestimates ecological richness (Tikhonov et al. 2015) and hence may end up with an incorrectly estimated number of OTUs/taxa.
Thus, some researchers began reconsidering the concept of OTUs and clustering-based OTU methods.
Some heuristic-based OTU methods have already tried to address the problem of how to choose an optimal distance threshold to define OTUs to represent different taxonomic levels. AGC (abundance-sorted greedy clustering) (Ye 2011) was developed for identifying and quantifying abundant species from pyrosequences of 16S rRNA by consensus alignment, aiming to avoid inflation of species diversity estimated from error-prone 16S rRNA pyrosequences. UPARSE (Edgar 2013) reported that the generated OTU’s sequences with ≤1% incorrect bases are more closer to the species in a community, and Swarm’s (Mahé et al. 2014) sub-OTUs solution is also an effort to refine the OTU clustering.
Both model-based and network-based clustering OTU methods have also been developed to address the problem of selecting optimal distance threshold to define OTUs (Wei et al. 2021).
Such model-based clustering OTU methods include CROP (Clustering 16S rRNA for OTU Prediction) (Hao et al. 2011), BEBaC (Cheng et al. 2012), and BC (Jääskinen et al. 2014). Such network-based clustering OTU methods include M-pick (Wang et al. 2013), (Wei and Zhang 2015), and DMclust (Wei et al. 2017).
However, within the framework of OTU methods, all these clustering methods including hierarchical, heuristic-based OTU methods and model-based and network-based OTU methods remain using the concept of OTUs; the difference between them is how to cluster sequences to OTUs and what cutoff values are used to refine OTUs as closer to species.
Until 2011, most researchers still believed that the solutions of OTU-based methods can be improved to quickly and accurately assign sequences to OTUs and hence the obtained taxonomic information from those OTUs can greatly improve OTU-based analyses. The OTU-based methods are considered being robust and can overcome many of the challenges encountered with phylotype-based methods (Schloss and Westcott 2011).
The first is to overcome the weakness of classical OTU approach based on an arbitrary sequence identity threshold because 97% similarity is not accurate and cannot approximate biology species.
The second is to overcome the amplicon sequencing errors. The classical OTU approach assumed that it could reduce problems caused by erroneous sequences. But it cannot solve the problem of erroneously clustering OTUs. It also reduces phylogenetic resolution because sequences below the identity threshold cannot be differentiated (Amir et al. 2017).
The third is to facilitate merging datasets generated by bioinformatic tools. Because the de novo OTUs were assessed having problem of merging OTUs and although this problem could be reduced in closed-reference and open-reference OTU picking (Rideout et al. 2014), it remains a challenge to integrate large datasets into a single OTU space (Amir et al. 2017).
The last two motivations are associated with downstream statistical analysis. To avoid these weaknesses, the new approaches focus on sub-OTU methods or algorithms to detect ecological differences by a single base pair and try to approve that the subunits are ecological and functional meaningfulness, while expecting sub-OTUs are feasible for analysis. The new direction of bioinformatic analysis of microbial community has been challenging the importance of the solution of OTUs and OTU-based methods. It aims to define atomic analysis units of microbiome data and to search for algorithms that can provide single-nucleotide resolution without relying on arbitrary percent similarity thresholds of OTUs (Eren et al. 2016) or refining the OTUs.
8.2.2 Single-Nucleotide Resolution Clustering-Based OTU Methods
Several computational methods or algorithms have been developed to improve the resolution of 16S data analysis beyond the threshold of 3% dissimilarity OTUs. We introduce the two most important algorithms below: distribution-based clustering (Sect. 8.2.2.1) and Swarm2 (Sect. 8.2.2.2).
8.2.2.1 Distribution-Based Clustering (DBC)
In 2013, Preheim et al. (2013) used DBC to refine the OTU for single-nucleotide resolution but without relying on sequence data alone to serve the dual purpose for the use of OTUs: identifying taxonomic groups and eliminating sequencing errors. Alternatively, the DBC method compares the distribution of sequences across samples. In other words, it uses ecological information (distribution of abundance across multiple biological samples) to supplement sequence information.
DBC algorithm is implemented under the framework of clustering OTU methods. It first uses Jukes-Cantor-corrected genetic distances or Jensen-Shannon divergence (JSD) as appropriate to measure the genetic distances among sequences and then implements either complete algorithm (all sequences are analyzed together in the analysis) or parallel algorithm (sequences are pre-clustered with a heuristic approach) or different algorithms (e.g., nearest-neighbor single-linkage clustering). DBC first uses the chi-squared test to determine whether two sequences have similar distributions across libraries. Then, based on ecological distribution to form OTUs, so 16S rRNA sequences that even differ by only 1 base but that are found in different samples are still put into different OTUs. Conversely, the sequences drawn from the same underlying distribution across samples are grouped together into the same OTU regardless of interoperon variation or sequence variation or random sequencing errors. The underlying assumption is that bacteria in different populations are often highly correlated in their abundance across different samples (Preheim et al. 2013). Thus, 16S rRNA sequences derived from the same population regardless of their variations or random sequencing error will have the same underlying distribution across sampled environments.
- 1.
DBC algorithm uses both genetic distance and the distribution of relative abundances of sequences across samples to identify the appropriate grouping for each taxonomic lineage and to detect many methodological errors. Thus, it may avoid the limitation that will necessarily either over-cluster or under-cluster sequences using genetic information alone.
- 2.
DBC method is more accurate and sensitive in identifying true input sequences, clustering sequencing, and methodological errors, in which the accuracy is in terms of the F-score and the Matthew’s correlation coefficient (MCC) (Baldi et al. 2000). The DBC method is more accurate (more correct OTUs, fewer spurious/incorrect OTUs) than other OTU methods including closed-reference (i.e., phylotyping), de novo clustering, and open-reference (i.e., a hybrid of phylotyping and de novo clustering) and UCLUST at grouping reads into OTUs. It is also sensitive enough to differentiate between OTUs that differ by a single base pair for showing evidence of differing ecological roles. In other words, DBC predicts the most accurate OTUs when sequences are distributed in an ecologically meaningful way across samples. It more accurately represents the input sequences based on the total number of OTUs and predicts fewer overall OTUs than the de novo and open-reference methods. Because detecting the differences between closely related organisms is crucial (Shapiro et al. 2012), thus, DBC can be chosen to distinguish the signal from the noise of sequencing errors and to form accurate OTU for identifying evolutionary and ecological mechanisms (Preheim et al. 2013).
- 1.
DBC is unreliable to conduct cross-sample comparisons for low-count sequences (Tikhonov et al. 2015).
- 2.
DBC currently requires very long run time on very large datasets and prohibitively long even for moderately sized datasets. This is a severe limitation of DBC (Preheim et al. 2013) and has been criticized (Tikhonov et al. 2015).
In summary, DBC is a clustering OTU method or a refined OTU method. The difference from other OTU methods is that it uses ecology to model multiple samples to refine the OTU. As a refined OTU method, DBC is still within traditional framework of OTU methods.
8.2.2.2 Swarm2
The de novo amplicon clustering methods share two fundamental problems (reviewed in Sect. 6.4). One uses arbitrary fixed global clustering thresholds; another is input-order dependency induced by centroid selection, i.e., the clustering results are strongly influenced by the input order of amplicons. To solve these two fundamental problems, in 2014, Mahé et al. (2014) proposed an amplicon clustering algorithm Swarm to fine-scale OTUs without relying on arbitrary global clustering thresholds and input-order dependency.
- 1.
Swarm was expected to reduce the influence of clustering parameters and to produce robust OTUs (Mahé et al. 2014).
- 2.
Swarm2 (Mahé et al. 2015b) directly integrated the clustering and breaking phases and hence improved the performances in terms of computation time and reducing under-grouping by grafting low abundant OTUs (e.g., singletons and doubletons) onto larger ones. The improvement is done through improving Swarm’s scalability to linear complexity and adding the fastidious option (Mahé et al. 2015b).
However, Swarm has the disadvantage: it cannot resolve at the single-base level (Hathaway et al. 2017).
Like distribution-based clustering (Sect. 8.2.2.1), Swarm and Swarm2 aim to provide single-nucleotide resolution (Mahé et al. 2014, 2015b). Other bioinformatic software that aim to single-nucleotide resolution include oligotyping (Section 8.3.1), denoising-based methods (Sect. 8.3.2) such as cluster-free filtering (Sect. 8.3.2.3), DADA2 (Sect. 8.3.2.4), UNOISE2 (Sect. 8.3.2.5), Deblur (Sect. 8.3.2.6), and SeekDeep (Sect. 8.3.2.7).
8.3 Moving Beyond the OTU Methods
We can use the following four characteristics to describe the beyond OTU methods: (1) to avoid the concept of OTUs, especially without relying on using 97% sequence identity to approximate species, (2) to avoid using clustering method, (3) to focus on single-nucleotide (a single base pair) resolution to provide more accurate estimates of diversity, and (4) to expect more clinical or functional relevancy. We use these four criteria especially the first three criteria to assess on whether or not the newly developed methods are moving beyond the traditional OTU methods.
8.3.1 Entropy-Based Methods: Oligotyping
In molecular biology, the term oligotyping mainly consists of dual DNA sequence analysis methods or functions of taxonomy and sequencing; that is, the approach of oligotyping is to use primary DNA sequencing to identify organism taxonomy (taxonomy), while improving the accuracy of DNA sequencing (sequencing).
In 2013, Eren et al. (2013) described a supervised computational method called oligotyping in analysis of 16S rRNA gene data and demonstrated that oligotyping is able to differentiate closely related microbial taxa. Oligotyping decomposes marker-gene amplicons with a different way from clustering OTUs. The basic theory underlying the oligotyping workflow is minimum entropy decomposition (MED) (Eren et al. 2014), the unsupervised oligotyping, which relies on Shannon entropy (Shannon 1948), a measure of information uncertainty (Jost 2006). MED is an algorithm for fine-scale resolution for Illumina amplicon data.
The oligotyping workflow is implemented via the following three procedures: First, MED identifies variable nucleotide positions among the entire sequencing data based on subtle nucleotide variation (Ramette and Buttigieg 2014). Second, MED only chooses those significantly variate positions to partition reads into oligotypes (“MED nodes”: representing homogeneous OTUs), i.e., sensitively partitioning the high-throughput marker-gene sequences (Eren et al. 2014). Third, MED produces a sample-by-OTU table for downstream analyses (Buttigieg and Ramette 2014).
- 1.
Could resolve closely related but distinct taxa that differ by as little as one nucleotide at the sequenced region.
- 2.
Could not only allow identifying nucleotide positions that likely carry phylogenetically important signal, and enable finer representing the microbial diversity in a wide range of ecosystems without relying on extensive computational heuristics and user supervision, but also improve the ecological signal for downstream analyses.
- 3.
Particularly, it was shown (Hathaway et al. 2017) that like SeekDeep (Sect. 8.3.2.7), MED was able to achieve 100% haplotype recovery using 454 and Ion Torrent pyrosequencing reads, as well as Illumina MiSeq reads on the Illumina platform.
However, it was also shown (Hathaway et al. 2017) that the MED algorithm appears to get trouble as a haplotype’s abundance increases.
8.3.2 Denoising-Based Methods
Typically many sequences generated by any sequencing platform contain at least one error (Amir et al. 2017). When microbiome researchers noticed that the sequences contain sequencing errors, the use of OTU methods has been changed (Gaspar 2018) to deal with sequencing errors.
Sequencing errors are extremely important confounding factors for detecting low-frequency genetic variants (Ma et al. 2019) and thus impact the underlying biology due to inaccurate taxon identification and inflated diversity statistics (Amir et al. 2017). Although these errors seldom affect statistical hypothesis testing of differences between two communities, clinically higher precision could be deviated by these errors (Amir et al. 2017). Removing or reducing the sequencing errors will reduce false-negative results and improve the accuracy of describing the microbial community. Thus, one direction of methodology development in bioinformatic analysis of microbiome data is to handle sequencing errors (Ma et al. 2019).
One way to deal with sequencing errors is to denoise them. Denoising-based or error-correction methods exploit the predictable structure of certain error types to attempt to reassign or eliminate noisy reads (Huse et al. 2010; Quince et al. 2011; Rosen et al. 2012). Most denoising algorithms aim to assign erroneous reads to their most likely source, to make the abundance estimates of true sequences more accurate (Tikhonov et al. 2015).
Several types of error are introduced by PCR amplification followed by sequencing (Edgar 2016b). Of which sequencing error, PCR single-base substitutions, and PCR chimeras are the three important sources of error (Quince et al. 2011): (1) point errors caused by substitution and gap errors due to incorrect base pairing and polymerase slippage, respectively (Turnbaugh et al. 2010), (2) PCR chimeras caused by extending an incomplete amplicon prime into a different biological template (Haas et al. 2011), and (3) spurious species due to contaminants from reagents and other sources (Edgar, 2013) as well as introduced when reads are assigned to incorrect samples due to cross talk (Carlsen et al. 2012).
Thus, to infer accurate biological template sequences from noisy reads, the denoising-based or error-correction methods typically take two steps (Edgar 2016b): first, denoising or correcting point errors to obtain an accurate set of amplicon sequences and then filtering the chimeric amplicons. Among them, DADA2, UNOISE3, and Deblur are the three most widely used denoising packages. We introduce them, respectively, as below.
8.3.2.1 Denoising-Based Methods Versus Clustering-Based Methods
- 1.
QIIME 2 outperformed MAPseq, mothur, and QIIME in terms of overall recall and F-scores at both genus and family levels as well as with the lowest distance estimates between the observed and simulated (predicted) samples, but at the expense of CPU time and memory usage. In contrast, MAPseq showed the highest precision, with miscall rates consistently <2%.
- 2.
Compared to using Greengenes, generally a higher recall was yielded using the SILVA database.
- 3.
The performance of taxonomic assignment for each tool can be considerably influenced varied up to 40% depending on the 16S rRNA subregion targeted. Overall, the V1–V2 and V3–V4 subregions performed the best across most of the software tools. However, the V1–V2 subregions were not recommended to be used for classification of complex community samples because the V1–V2 primers did not match almost 70% of the sequences across the four reference databases.
- 4.
This study concluded that use of either QIIME 2 or MAPseq is optimal for 16S rRNA gene profiling, where the recall rate (sensitivity) was estimated as the percentage of sequences assigned to the expected taxa for each biome, while precision (specificity) was calculated as the fraction of sequences from these predicted taxa out of all those from the taxa observed. The distances were estimated with either the Bray-Curtis or Jaccard dissimilarity indices at the genus level. The F-score was calculated as
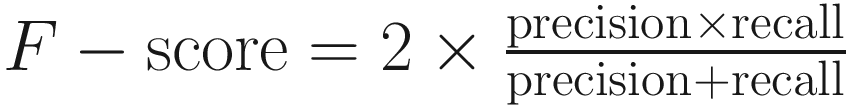 , where precision is defined as
, where precision is defined as 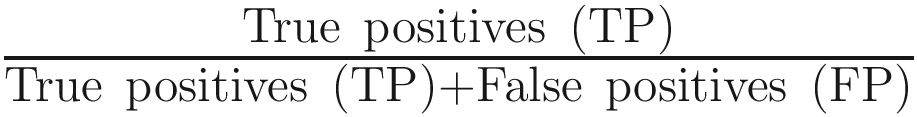 and recall is defined as
and recall is defined as 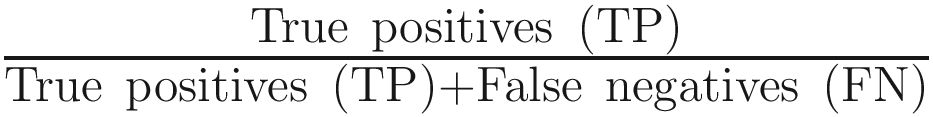 .
.
- 1.
The mock community analyses showed that these three denoising pipelines generate very different numbers of ASVs that significantly impact alpha diversity metrics although they produced similar microbial compositions based on relative abundance.
- 2.
The real analyses showed that the three packages were consistent in their per-sample compositions, resulting in only minor differences of the intra-sample distances based on weighted UniFrac and Bray-Curtis dissimilarity.
- 3.
Overall, open-reference OTU clustering approach consistently identified considerably more OTUs than the number of ASVs generated by the denoising pipelines in all datasets tested.
- 4.
It was shown that DADA2 tended to find more ASVs than UNOISE3 and Deblur (Deblur called the least amount of ASVs), suggesting that it could be better at finding rare organisms, but at the expense of possible false positives.
- 5.
It also showed that UNOISE3 is very faster than DADA2 and Deblur, with DADA2 being the lowest with 1200 times lower than UNOISE3.
- 6.
This study concluded that all pipelines result in similar general community structure; however, the number of ASVs/OTUs and resulting alpha diversity metrics varies considerably. Thus when attempting to identify rare organisms from possible background noise, we should consider that determining species richness within low-diverse samples could be problematic for the denoising pipelines (Nearing et al. 2018).
- 1.
DADA2 offered the best sensitivity but at the expense of decreased specificity compared to USEARCH-UNOISE3 and Qiime2-Deblur.
- 2.
USEARCH-UNOISE3 had the best balance between resolution and specificity (Prodan et al. 2020).
- 3.
Compared to ASV-level software, OTU-level software USEARCH-UPARSE and mothur performed well, but with lower specificity, whereas QIIME-uclust produced large number of spurious OTUs as well as inflated alpha diversity measures.
8.3.2.2 Pyrosequencing Flowgrams
PyroNoise (Quince et al. 2009, 2011). The PyroNoise algorithm is the first amplicon sequencing error-correction method that was designed for clustering the flowgrams of 454 pyrosequencing reads using a distance measure to model sequencing noise. PyroNoise is used to accurately construct OTUs from 16S rRNA sequence data and assign pyrosequenced reads to known taxa (Quince et al. 2009). It may be more robust to noise. However, PyroNoise is computationally expensive and is difficult to implement for most users and thus has received limited application (Schloss et al. 2011).
DeNoiser (Reeder and Knight 2010) is another denoising pyrosequencing algorithm that was proposed for denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Like PyroNoise, DeNoiser was reviewed as computationally expensive and difficult for most users and has limited application (Schloss et al. 2011).
SLP (single-linkage pre-clustering) (Huse et al. 2010) was proposed to address the problem that the complete-linkage clustering significantly increases the number of predicted OTUs and inflates richness estimates. SLP implements its algorithm via four steps: First it orders the unique sequences by frequency. Second, from the most abundant to the least sequence, it assigns them to clusters through testing each subsequent sequence against the growing list of clusters using the single-linkage algorithm. Third, it adds a new sequence to the cluster and stops testing against subsequent clusters if the sequence has a pairwise distance less than 0.02 (2%) to any of the sequences already in the cluster. It was shown that SLP can more accurately predict expected OTUs if followed by an average-linkage clustering based on pairwise alignments (Huse et al. 2010) and is not computationally expensive (Schloss et al. 2011).
AmpliconNoise (Quince et al. 2011), a development of the PyroNoise algorithm, was proposed to remove noise from pyrosequenced amplicons to avoid inflation of diversity estimates of OTUs. It uses the error-model-based denoising algorithm to distinguish noise from true sequence diversity in data and uses expectation-maximization (EM) to inference the true sequences. It was shown that AmpliconNoise can separately remove 454 sequencing errors and PCR single-base errors to obtain accurate estimates of OTU number (Quince et al. 2011).
APDP (Amplicon Pyrosequencing Denoising Program) (Morgan et al. 2013) was proposed to process raw sequence datasets into a set of validated sequences with compatible formats for facilitating downstream analyses. APDP uses both between-samples abundance distribution of similar sequences and within-samples frequency and diversity of sequences to distinguish real sequences from errors generated by PCR and sequencing. It was shown that APDP can effectively remove errors from both deeply sequenced datasets comprising biological and technical replicates and efficiently denoise single-sample datasets and thus provides more conservative or accurate biological diversity estimates (Morgan et al. 2013).
Other 454 sequence programs for correcting pyrosequencing errors include Acacia (Bragg et al. 2012) and HECTOR (Wirawan et al. 2014).
Compared to denoising pyrosequencing flowgrams, Illumina denoisers have been developed more recently. Some Illumina methods can also be used with the 454 sequencing platform, such as cluster-free filtering (CFF) and SeekDeep. Below we will introduce CFF in Sect. 8.3.2.3, DADA2 in Sect. 8.3.2.4, UNOISE2 in Sect. 8.3.2.5, Deblur in Sect. 8.3.2.6, and SeekDeep in Sect. 8.3.2.7.
8.3.2.3 Cluster-Free Filtering (CFF)
In 2015, Tikhonov et al. (2015) proposed a cluster-free filtering (CFF) denoiser approach to achieve sub-OTU resolution with ecologically differing by as little as one nucleotide (nt) (99.2% similarity). The CFF denoiser was proposed in the contexts: on the one hand, to improve estimates of ecological diversity by reducing the impact of amplicon sequencing error, researchers have tried various denoising algorithms to reassign or eliminate noisy reads; however, the challenging still remains because no approach can fully address the denoiser issues. On the other hand, the alternative DBC approach is necessarily unreliable due to computational cost and severe limitation for low-count sequences in the cross-sample comparisons. Under this background, the focuses of the CFF denoiser are not to further try to improve the existing OTU clustering approaches and also not to attempt to identify rare species. Instead, the CFF denoiser combines error-model-based denoising and systematic cross-sample comparisons to resolve the fine (sub-OTU) structure of moderate-to-high-abundant taxonomy data (Tikhonov et al. 2015).
CFF employs cross-sample comparisons to achieve sub-OTU resolution. CFF was developed based on the observation that sequence similarity need not imply dynamical similarity and vice versa in time-series data. In contrast, the maximum correlation between the time traces (normalized counts versus observation day) of two sequences depends on their abundance measured by Poisson model. Thus CFF denoiser (Tikhonov et al. 2015) first defines the “dynamical similarity” of two traces as the Pearson correlation of their abundance, normalized by their maximum possible correlation. Then it models the abundance traces of these two sequences by an additive Poisson counting noise, setting an upper bound on the correlation coefficient for low-abundance sequences because Poisson sampling noise becomes non-negligible for low-abundance sequences. Finally it uses the Hamming distance metric to measure the pairwise sequence distances based on normalized counts.
Three important characteristics that characterize CFF as a method of moving beyond the traditional OTU methods are as follows: (1) aims to differ 16S sequences by one nucleotide or a single base pair, which is similar to oligotyping and distribution-based clustering approaches; (2) does not rely on clustering similar sequences together, which is similar to oligotyping; and (3) does not assume that sequence similarity implies ecological similarity. Instead CFF actually considers sequence similarity is a very poor predictor of ecological similarity (Tikhonov et al. 2015). Thus CFF uses highly conservative filtering criteria, assuming that sequence similarity contains only true biological sequences, that is, there are no false positives. Regarding this point, CFF is similar to Sokal and Sneath’s numerical taxonomy (Sokal and Sneath 1963).
- 1.
CFF demonstrated “sequence similarity need not imply ecological similarity, and vice versa,” and the ecological relatedness can be independently assessed. This is different from the tag-sequencing data analysis and clustering-based OTU methods in microbiome study because typically these methods assume that sequence similarity of 16S hypervariable regions can be used as a proxy for phylogenetic, and therefore ecological, relatedness (Tikhonov et al. 2015). This idea of CFF is similar to the important idea of separating taxonomic procedure from phylogenetic speculation in the original OTU methods (Sokal and Sneath 1963).
- 2.
CFF can achieve a single-nucleotide (nt) sub-OTU resolution (Tikhonov et al. 2015).
- 3.
Particularly, CFF can achieve the same accuracy as DADA, which was considered as the best denoiser in 2015 (Tikhonov et al. 2015), but has less execution time than DADA. We can ignore the advantage of execution time since the longer time used in DADA largely due to its exact treatment of probabilities for processing low abundant sequences while CFF denoiser discards sequences of low abundance prior to denoising. In other words, the speed of CFF is at the cost of its limitation.
However, CFF also has limitations: (1) It is specifically designed to be run on large multi-sample datasets. (2) It is not a replacement for OTU clustering; it focuses on moderate-to-high-abundance sequences and hence discards low-abundance sequences. Thus, CFF is unsuitable for studying population-level alpha or beta diversity (Tikhonov et al. 2015), which probably is an important limitation of this method.
8.3.2.4 DADA2
DADA2 and q2-dada2 plugin were introduced in Sect. 4.2.1. In line with providing single-nucleotide resolution without using arbitrary taxonomic similarity thresholds to define molecular OTUs, Callahan et al. (2017) proposed to use exact “sequence variants” to replace OTUs in marker-gene data analysis and developed software package DADA2 (DADA: Divisive Amplicon Denoising Algorithm) for modeling and correcting Illumina-sequenced amplicon errors. The proposal of using ASVs instead of the traditional OTUs was due to the limitations of OTU methods, such as clustering sequence reads into OTUs often eliminates biological information present in the data, and OTUs are not species, and their construction is not necessitated by amplicon errors (Callahan et al. 2016a), and especially was motivated by a series of new computational methods (Eren et al. 2013, 2016; Tikhonov et al. 2015; Edgar 2016b; Amir et al. 2017).
DADA2 is a parametric model that relies on input read abundances and distances. It assumes that true reads are likely to be more abundant and less abundant reads are likely error-derived which may be only a few base differences away from a more abundant sequence (Prodan et al. 2020; Callahan et al. 2016a). DADA2 was developed based on the DADA (Rosen et al. 2012). DADA was built on AmpliconNoise’s error-modeling approach (Quince et al. 2011). The distinctive feature of DADA is its divisive hierarchical clustering algorithm, while previous methods, including AmpliconNoise and simple OTU clustering, use agglomerative approach.
The core denoising algorithm (Divisive Amplicon Denoising Algorithm) in the DADA2 is based on a Poisson model to estimate the errors in Illumina-sequenced amplicon reads. This error model estimates the probability for each amplicon read with each sequence is produced from sample sequence as a function of sequence composition and quality. The Poisson model assumes that the number of amplicon reads (the abundance) of each sequence is consistent with the error model and the p-value of the null hypothesis is calculated. The calculated p-values or quality scores (base Q-scores) are used as the division criteria for an iterative partitioning algorithm; sequencing reads will continue to be divided until all partitions are consistent with being produced from their central sequence.
- 1.
DADA2 builds unique error models for each sequencing run (Nearing et al. 2018; Callahan et al. 2016b).
- 2.
DADA2 exactly infers sample sequences and resolves differences of as little as one nucleotide (Callahan et al. 2016b).
- 3.
DADA method was shown to outperform AmpliconNoise’s and other previous methods in both speed and accuracy (Rosen et al. 2012).
- 4.
DADA2 R package can implement the full amplicon workflow from filtering, dereplication, sample inference, and chimera identification to merging the paired-end reads (Callahan et al. 2016a). Inferring ASVs (also called RSVs: ribosomal sequence variants) was shown in amplicon bioinformatic workflow (Callahan et al. 2016a, b, 2017).
- 5.
Particularly, it was demonstrated that the DADA2 methods were more accurate than the four OTU methods: UPARSE (Edgar 2013), MED (minimum entropy decomposition) (Eren et al. 2015), mothur (average linkage) (Schloss et al. 2009), and QIIME (uclust) (Caporaso et al. 2010).
- 1.
As reviewed above, it was shown (Nearing et al. 2018; Prodan et al. 2020) that DADA2 had the best sensitivity and resolution, but at the expense of producing more number of spurious ASVs compared to USEARCH-UNOISE3 and Qiime2-Deblur.
- 2.
It was also shown (Hathaway et al. 2017) that like MED and UNOISE, DADA2 could create larger false haplotypes especially for 454 technique. In DADA2, when the number of false haplotypes is minimized, the sensitivity is also lost, especially for the lower read depth input, in which the low-abundance one-off haplotypes are missed (Hathaway et al. 2017). For Ion Torrent and 454 pyrosequencing data, both UNOISE and DADA2 cannot achieve 100% haplotype recovery compared to 100% haplotype recovery in SeekDeep and MED (Hathaway et al. 2017).
- 3.
It was also shown (Tikhonov et al. 2015) that DADA has a long execution time largely due to its exact treatment of probabilities. This is critically important to process sequences with an abundance of just a few counts.
Overall, DADA2 has been recognized as so far the best denoising-based method for focusing on the highest possible biological resolution (e.g., differentiating closely related strains) (Prodan et al. 2020).
8.3.2.5 UNOISE2 and UNOISE3
To improve error correction in Illumina 16S and ITS amplicon sequencing, distinguishing the sequence errors caused by PCR and sequencing from true biological variation, Edgar (2016b) proposed UNOISE2, an updated version of the UNOISE algorithm (Edgar and Flyvbjerg 2015) for denoising (error-correcting) Illumina amplicon reads.
UNOISE2 uses a one-pass clustering strategy without using quality (Q) scores. The clustering strategy is different from DADA2’s model-based approach, which uses quality scores in an iterative divisive partitioning clustering strategy. In UNOISE2, both algorithms of UCHIME2 (Edgar 2016a) and DADA2 are incorporated to reduce the number of incorrect sequences. However, both UNOISE2 and DADA2 were reviewed as still generating some amount of false positives with DADA2 likely to increase the number of false-positive chimera predictions (Edgar 2016b). In UNOISE2, the obtained predicted biological sequences are called as ZOTUs (zero-radius OTUs) (Edgar 2016b, 2018). The ZOTU is similar to Callahan et al.’s ASV. In DADA2, the 97% similarity of OTUs is replaced by ASVs as the standard unit of marker-gene analysis and reporting; in UNOISE2, similarly the [97%] OTUs are replaced by ZOTUs (Edgar 2018).
The UNOISE3 denoising method first uses the “unoise3” command to rank sequences in decreasing order of abundance and then discards those sequences with counts less than the specified minimum abundance threshold (the default value is 8). By using 100% identity threshold, each distinct sequence defines a separate OTU. Both OTU-level clustering with UPARSE and ASV-level denoising with UNOISE3 are implemented via USEARCH, and actually the author of USEARCH (Edgar 2010) recommended that UPARSE and UNOISE3 should be performed together.
- 1.
UNOISE2 has comparable or better accuracy than DADA2 demonstrated by the author of UNOISE2 (Edgar 2016b).
- 2.
USEARCH-UNOISE3 (UNOISE2) had arguably the best overall performance compared to DADA2 and Qiime2-Deblur if combinedly considering its high sensitivity with excellent specificity (Prodan et al. 2020).
- 3.
Especially USEARCH-UNOISE3 and Qiime2-Deblur had the perfect specificity, producing no spurious OTUs/ASVs based on the mock sample sequencing data analysis (Prodan et al. 2020).
- 1.
Like DADA2, UNOISE was designed and tested on Illumina reads. It does not work well on Ion Torrent and 454 data and hence cannot achieve 100% haplotype recovery (Edgar 2021; Hathaway et al. 2017).
- 2.
UNOISE collapses one-off errors if the abundance ratio of two sequences achieves a certain threshold, recommends not using singlet sequences, and hence decreases haplotype recovery at lower read depths. This in part contributes to UNOISE’s speed and makes it to be the fastest algorithm compared to MED, DADA2, and SeekDeep (Hathaway et al. 2017). However, this also costs UNOISE to be unable to detect new haplotypes that differ by only one nucleotide (Hathaway et al. 2017).
8.3.2.6 Deblur
We illustrated Qiime2-Deblur in Chap. 4 (Sect. 4.4). Amir et al. (2017) developed a novel sub-OTU (sOTU) method called Deblur to fast and accurately identify exact amplicon sequences and to integrate large datasets. Like DADA2 and UNOISE2, Deblur aims to identify real ecological differences between taxa with a single base pair of amplicons differentiation. Deblur in concept is similar as DADA2 and UNOISE2, but uses a different algorithm to obtain single-nucleotide resolution. Deblur performs each sample independently and compares sequence-to-sequence Hamming distances within a sample to an upper-bound error profile by combining with a greedy algorithm (Amir et al. 2017).
The Deblur algorithm is implemented via three steps (Amir et al. 2017): First, Deblur sorts sequences by abundance. Second, Deblur subtracts the number of predicted error-derived reads from the counts of neighboring reads based on their read-to-read Hamming distance using an upper bound on the error probability based on the most to least abundant sequence. Finally, Deblur removes any sequence whose abundance drops to 0 after a subtraction. Thus, after applying Deblur, the invalid (i.e., noise) sequences are removed, and only reads likely to have been presented to the sequencer are retained.
- 1.
Deblur is comparable or better than DADA2 and UNOISE2 in terms of performance characteristics and stability (i.e., obtaining the same sOTU across different samples) (Amir et al. 2017). For example, although all three methods can identify sOTUs with single-nucleotide differences which are close to the ground truth, Deblur has the greater stability than DADA2 and UNOISE2.
- 2.
Unlike DADA2 and UNOISE2, Deblur performs each sample independently without requiring operation on the full study and therefore can be easily parallelized to very large projects.
- 3.
Particularly, in one study that compared six bioinformatic software for the analysis of amplicon sequence data including QIIME-uclust, mothur, and USEARCH-UPARSE for generating OTUs and DADA2, Qiime2-Deblur, and USEARCH-UNOISE3 for generating ASVs, Qiime2-Deblur is one of the only two pipelines (another is USEARCH-UNOISE3) that showed perfect specificity on the mock sample sequencing data, producing no spurious OTUs/ASVs (Prodan et al. 2020).
Deblur also has the disadvantages, such as Deblur has poor performance compared to DADA2 and open-reference OTU clustering when it is used to detect low abundant taxa in the extreme dataset at 97% identity (Nearing et al. 2018). For comparing Deblur and DADA2, the reader is also referred to Sect. 4.4.6.
8.3.2.7 SeekDeep
SeekDeep (Hathaway et al. 2017) is an error-correction method for de novo (i.e., reference-free) analysis of amplicons. SeekDeep was developed in the contexts of comparing to three pipelines that aim for single-base resolution to determine the local PCR amplicon haplotypes (simply as haplotypes for brevity), MED (Eren et al. 2014), DADA2 (Callahan et al. 2016a), and UNOISE in USEARCH (Edgar 2016b), as well as two OTU-based clustering pipelines which cannot resolve at the single-base level: USEARCH (aka UCLUST/UPARSE) (Edgar 2016b) and Swarm (Edgar 2013). SeekDeep aims for single-base resolution to provide improved resolution toward species and strains. The central algorithm of SeekDeep is the qluster (for quality clustering) that improves the correction of PCR and sequencing errors through base quality values and k-mer frequencies and other multiple key ways. SeekDeep can be utilized with Ion Torrent, 454, and Illumina sequencing technologies. SeekDeep consists of four components (Mahé et al. 2014): (1) extractor for de-multiplexing and read filtering, (2) qluster for rapid and accurate clustering based on quality, (3) processClusters for replicate and population comparisons, and (4) popClusteringViewer for viewing and manipulating final results. The first three main components are central to generating clustering results, and the fourth is an additional component to aid in viewing and sharing the results.
- 1.
SeekDeep is able to resolve sequences differing by only a single base.
- 2.
SeekDeep has greater consistency even at low frequencies.
- 3.
Particularly, SeekDeep has matched or outperformed the five pipelines including MED, DADA2, UNOISE in USEARCH, USEARCH, and Swarm in terms of haplotype recovery (especially one-off haplotypes), accuracy of predicting the abundance (accurate abundance estimates), and the number and abundances of false haplotypes.
However, Hathaway et al. (2017) also acknowledged that SeekDeep creates more false haplotypes than DADA2 and UNOISE although the false haplotypes have much lower abundance.
8.3.2.8 Remarks on Denoising-Based Methods
Denoising-based methods are widely used for identifying low-abundance or rare species against a noisy background, often with the aim of improving estimates of ecological diversity (Hathaway et al. 2017), such as Chao1 richness.
Using 97% identity may merge phenotypically different strains with distinct sequences into a single cluster (Tikhonov et al. 2015; Callahan et al. 2016a, b, c). In contrast, the denoising usually results in a set of predicted biological sequences or valid OTUs, which often have single-base resolution. Thus, compared to OTU methods, in most cases denoising algorithms provide the maximum possible biological resolution and hence superior to conventional OTU methods.
However, the denoising-based methods cannot fully address all the issues of bioinformatic analysis of microbiome data although these methods were proposed for identifying rare species and improving the accuracy of ecological diversity estimates, because (1) all error models are necessarily approximate, and no denoising algorithm can deal with errors that are beyond their model capabilities, and (2) the denoising-based methods are particularly problematic when they are used for calling low-abundance species. As reviewed above, DADA2 had the best sensitivity and resolution, but also produced more number of spurious ASVs, while UNOISE2 was fast, but was not recommended for using singlet sequences due to decreased haplotype recovery at lower read depths.
8.4 Discussion on Moving Beyond OTU Methods
The concept of OTU and OTU methods were originated from numerical taxonomy. OTU is an operational taxonomic unit or the organizational level of a unit character. It was created in numerical taxonomy to facilitate taxonomic classification against previous subjective approaches of taxonomy. The OTU methods in numerical taxonomy can be characterized with three concepts and one assumption. The three concepts are OTU, similarity, and clustering. The assumption is that taxonomic classification can be done through clustering the similarities of characters with large random sampling. It separates taxonomic classification from phylogenetic speculation. In numerical taxonomy, the OTUs are used for classification of characters via commonly used clustering with the assumption that characters can be grouped based on their similarities.
In early microbiome studies, the concept of OTU and OTU methods were adopted for convenience, and using the 97% sequence identity for approximating species is arbitrary and was based on researchers’ experiences but not substantial experiments. In microbiome, the use of OTUs serves the dual purpose: grouping sequences (the third purpose of playing species functionality is the extension of the first purpose) and estimating diversity. The final purpose of using OTUs is to detect the association between microbiome and health and the development of the disease. When the concept of OTU and OTU methods were adopted into microbiome study, not only the commonly used clustering methods but also various methods including heuristic clustering, model-based methods were developed to improve and refine OTUs, all with the assumption that sequence similarity can predict taxonomic similarity.
Under the framework of OTU methods, the dual purpose of using OTUs has been demonstrated failing to achieve. In the meanwhile, various new methods have been proposed in recent years with the goal to achieve single-nucleotide resolution of sequences and not necessarily relying on arbitrary sequence identity and clustering methods. Among different methods that were newly proposed, the denoising-based methods have been demonstrated their promising. However, either the failure of dual purpose of using OTU methods or the promising of new bioinformatic sequence analysis raises a number of interesting questions which deserve further discussion.
8.4.1 Necessity of Targeting Single-Base Resolution
First, whether it is necessary to target single-base resolution?
On the one hand, clinically and for downstream analysis of microbiome data, a consistent single-base resolution of analysis units is necessary. Clinically, consistently differentiating single-base differences of analysis units is particularly a necessity for studying eukaryotic intraspecies populations and for mutation detection. It is crucial for seeking to detect and quantify minority haplotypes that may be represented by a single-nucleotide polymorphism (SNP), a nucleotide difference in a single DNA building block. SNP is the most common type of genetic variation among people. SNPs have been used to detect strains (also refers to subspecies) of clinical relevance or to predict phenotypic characteristics when they are stably associated with other parts of the bacterial haplotype (Fitz-Gibbon et al. 2013). In the oncology and infectious disease fields, such studies are becoming increasingly common. In these studies, these sequences often only differ from the wild type by a single base when using marker regions to bacterial strains or monitoring for pathogen drug resistance mutations. Thus, it is crucial to accurately quantify these low-abundance and genetically similar strains (Miotto et al. 2015; Hathaway et al. 2017). Another example provided by Tikhonov et al. (2015) is malaria research, where the strains within an infected individual are often defined by the sequence of a single amplicon, and these sequences usually differ by only a single base, representative of a SNP within the larger parasite population.
Recently, Johnson et al. (2019) suggested that intragenomic variation between 16S gene copies must be necessarily accounted for in modern bioinformatic analysis. They also demonstrated that full-length 16S intragenomic copy variants can potentially provide taxonomic resolution of bacterial communities at species and strain level when they are appropriately treated, where the full 16S gene refers to the ~1500 bp 16S rRNA gene comprising nine variable regions (V1–V9) interspersed throughout the highly conserved 16S sequence. Hassler et al. (2022) found that alignment SNP count strongly predicted concordance for any given gene and the strongest concordance was displayed by the SNPs from non-ribosomal protein coding genes and the weakest concordance was shown by the SNPs from rRNA genes. Statistically, single-base resolution of 16S amplicon clustering improves accuracy and sensitivity of traditional OTUs and thus extracts maximal information for downstream analysis (Tikhonov et al. 2015).
On the other hand, the techniques for obtaining a consistent single-base resolution of atomic analysis units are still challenging. For example, as we reviewed, DADA2 could have the best sensitivity and resolution, but also is prone to inflate spurious ASVs, while UNOISE2 could be fast, but without recommending using singlet sequences because of decreased haplotype recovery at lower read depths. SeekDeep may create more false haplotypes than DADA2 and UNOISE2.
8.4.2 Possibility of Moving Beyond Traditional OTU Methods
Second, whether it is possible to move beyond the traditional OTU-based methods?
Regardless of the names either called “oligotypes,” “ASVs,” “ZOTUs,” “sOTU,” or others, the authors of all the newly developed methods explicitly or inexplicitly want to replace the traditional OTUs by their proposed sub-OTUs.
- 1.
The new available methods provide better resolution than OTU methods. The sub-OTU methods can obtain single-nucleotide resolution, which makes it possible to identify real ecological differences between taxa or organisms in communities whose amplicons differ by a very small number of nucleotides (i.e., a single base pair, a single nucleotide) (Hathaway et al. 2017; Eren et al. 2013; Callahan et al. 2016a; Amir et al. 2017). For example, the identified oligotypes by oligotyping offer a comparable or higher level of resolution than OTUs at 97%, while the traditional OTU clustering at 97% identity level does not have so high resolution.
- 2.
The new available methods are more accurate than OTU methods with demonstrating as good or better sensitivity and specificity than OTU methods. For example, oligotyping and ASV methods can separate different environments more efficiently and have increased power to discriminate better ecological distribution patterns that taxonomical classification and OTU clustering at 97% identity level cannot detect (Eren et al. 2013, 2014, 2015; Callahan et al. 2016a). ASVs even can capture all biological variation present in the data (Callahan et al. 2017). Swarm2 can distinguish higher-resolution clusters or different taxa, while traditional UCLUST cannot detect (Callahan et al. 2017). Deblur also has similar or better sensitivity and specificity while it substantially reduces computational demands relative to similar sOTU methods (Mahé et al. 2015b).
- 3.
Like PiCRUST (Amir et al. 2017), sub-OTUs are functional and biological or clinically meaningful. For example, the identified subpopulations of a single species by a sequence or the sub-OTU can provide functional information: insight into functional relatedness of community members (Langille et al. 2013). Sequences differing by as little as one nucleotide (99.2% similarity) within the 16S rRNA gene can be ecologically distinct and reveal clinical or epidemiological associations that would be missed by genus-level or species-level categorization of 16S rRNA data (Tikhonov et al. 2015).
- 4.
The sub-OTUs facilitate downstream analysis due to their precision, reusability across studies, and reproducibility in future datasets. The sub-OTU methods, such as oligotyping, cluster-free filtering, and DADA2, were shown not only comparable or better than traditional clustering analysis at explaining the structure of the bacteria dataset but also can avoid vast underestimates of ecological richness, improve accurate measurement of diversity and dissimilarity, provide new insight into factors shaping community assembly and the prevalence of strain exchange between communities and invasion/extinction dynamics of OTU subpopulations, and resolve distinct subpopulations with high dynamical similarity. Thus, due to its precision, the sub-OTU methods could potentially allow amplicon methods to probe strain-level variation (Eren et al. 2011, 2013; Callahan et al. 2016a). For traditional OTU methods, after filtering the sequences and removing the chimer, a sample-by-OTU feature table is generated to serve as the basis for further analysis. Most sub-OTU methods also can obtain a sample-by-sub-OTU feature table for downstream analysis. Probably, more importantly, sub-OTUs are reusable across studies and comprehensive and reproducible in future datasets because they are more precise and combine the benefits for subsequent analysis of closed-reference and de novo OTUs (Tikhonov et al. 2015). We can continue our discovery by exploring the present and future large datasets and through reuse of existing rich datasets (Callahan et al. 2017). Some sub-OTUs, such as DADA2, can easily process Illumina samples on a laptop, and a workflow from raw data to downstream analysis was shown in recent publications (Callahan et al. 2016a, 2017; Amir et al. 2017). In summary, all these advantages of sub-OTUs, especially more accurate abundance estimates of sub-OTUs, facilitate downstream statistical analyses using sophisticated parametric or nonparametric models (Callahan et al. 2016b).
Based on above review, the sub-OTU methods are better than OTU methods. Most of them are stable and able to integrate and have good performance and open-source license. Using sub-OTU methods in marker-gene data analysis will be a trend in bioinformatic analysis of microbiome data or at least is an alternative method of OTU approach. The sub-OTUs are the good alternative to OTUs as the analysis units of microbiome data.
The usage of these new sub-OTU methods is facilitated by using them in datasets and bioinformatics pipelines. For example, since its introduction, Swarm v1 has been used in a variety of datasets (Callahan et al. 2016a; de Vargas et al. 2015; Filker et al. 2015; Lima-Mendez et al. 2015; Mahé et al. 2015a). QIIME already offers Swarmv1.2, to facilitate its usage, and Swarm v2 can be included in QIIME2 and in Galaxy (Oikonomou et al. 2015; Mahé et al. 2015b). QIIME 2 has already incorporated DADA2 by the use of exact “ASVs” rather than “OTUs.” Because of its stability and open-source license, Deblur is easy for commercial adoption and peer scrutiny and to be integrated (Goecks et al. 2010). The R package DADA2 was developed to show its usage in analysis of microbiome data (Amir et al. 2017).
8.4.3 Issues of Sub-OTU Methods
Third, whether there still exist issues in sub-OTU methods after moving beyond the use of OTUs and OTU-based methods?
Sub-OTU methods open potential new directions for a more accurate depiction of microbiome communities through marker-gene amplicons. However, the new methods also have the issues and come with new questions.
First, although the new methods can distinguish highly similar sequence variants, there is still no guarantee that the resulting sub-OTUs are necessarily phylogenetically and ecologically meaningful due to limitations of the selected 16S rRNA gene (Callahan et al. 2016a; Berry et al. 2017). Thus, the inference based on sub-OTUs (i.e., ASVs) does not solve all problems (Callahan et al. 2017). Second, not all new methods are suitable for studies of microbiome communities. For example, in the cluster-free filtering, the low-abundance sequences are discarded; thus, we cannot use it to study population-level alpha or beta diversity (Callahan et al. 2017). Third, to be more accessible, computational and ecological issues still need to be addressed for new methods. To move beyond the use of OTUs, the microbial ecologists and the developers of widely used software platforms still need further works (Tikhonov et al. 2015).
8.4.4 Prediction of Sequence Similarity to Ecological Similarity
Fourth, whether we need to avoid the assumption that the sequence similarity predicts the ecological similarity or phylogeny?
In literature, on the one hand, it was exhibited that 16S-based methods have limitations, and on the other hand, it was demonstrated that even a purely 16S-based study can exhibit ecologically significant distinctions to provide insight into functional relatedness of community members (Eren et al. 2016; Langille et al. 2013; Tikhonov et al. 2015). Thus, the assumption of the sequence similarity predicting the ecological similarity or phylogeny remains unanswered.
8.4.5 Functional Analysis and Multi-omics Integration
Fifth, whether functional analysis and multi-omics integration are necessary and important in microbiome study?
Discussion on the advantages and disadvantages of the OTU methods and the movement of moving beyond OTU methods highlights the importance of two topics in bioinformatic and statistical analysis of microbiome data: (1) What are the optimal analysis units of microbiome data? (2) What kind of science microbiome should be?
Regarding the first topic, compared to OTU methods, in general the methods that provide ASVs or single-nucleotide base resolution are more close to microbiome sequence data and serve the purposes of microbiome study. We have shown that targeting single-base resolution is necessary and the movement of moving beyond traditional OTU-methods is possible in bioinformatic analysis of microbiome data. However, as we reviewed above, the ASVs and single-nucleotide base resolution methods also have limitations, and not all OTU methods are inferior to all the ASV methods. Actually, all statistical models are used to approximate the real data but are not expected to get insight into all the complexities of reality; i.e., every single model will never represent the exact real data and rare achieve all the purposes of the analysis and the study supposed to be; the goal of model comparisons is to find the model that is sufficiently close to the reality because it is useful even if this model cannot describe exactly the reality. The challenge is how to choose the parsimonious bioinformatic models that provide remarkably useful approximations to the real sequencing data and extract maximal information for statistical analysis to obtain insight into clinical relevance. Here, Ockham’s razor [William of Ockham (c. 1287–1347)] or the principle of parsimony or law of parsimony is applicable. The principle of parsimony states that we should choose the simplest model while approximating to reality because “entities should not be multiplied beyond necessity” (Schaffer 2015).
Regarding the second topic: what kind of science is microbiome? These are the questions: How to define microbiome? What research theme in microbiome study? What are the unique data structure and characteristics? The interested reader is referred to Xia and Sun (2022).
Briefly, we define microbiome as an experimental science. As an experimental science, microbiome does not solely lie on direct observation and measurement of objects/samples and phenomena to identify association or correlation, but more importantly conducts experiments to find the functions and mechanism/causality that play in the human health and the development of disease. Like other experimental sciences, “inference from the particular to the general must be attended with some degree of uncertainty, but this is not the same as to admit that such inference cannot be absolutely rigorous, for the nature and degree of uncertainty may itself be capable of rigorous expression” [see Fisher (1935)’s Design of Experiments (p. 4)].
Actually, for microbiome study, the accurate coding of sequence data by a bioinformatic tool is just the first step. We need multiple approaches, especially including experiments and clinical trials to conform and validate the findings of bioinformatic analyses.
Microbiome study as a research field is not merely descriptive and observational. Describing diversity and visualizing the differences between the experimental groups are necessary. But the final goal of microbiome study is to find the causality between microbiome and the human health and the development of disease and to improve human health and prevent disease using the findings from microbiome research. The diversity concept was adopted from macroecology. The alpha diversity including richness and evenness are important in macroecology. But are they equally important in microbial ecology if considering their differences and complicities of microbial ecology? The answer is no.
Currently two approaches in microbiome research tend to be more important although they are more complicated: One is functional study, which is to find out the functionality of microbiota, identify the top microbes, and use them in experiment study to test their functions. Another is to integrate microbiome with other omics, such as metabolomics, metaproteomics, transcriptomics, genomics, and epigenomics to find the biomarkers that are associated with favorable and adverse outcomes. Finally the findings from microbiome research are expected to apply to human health and treat the human diseases.
Overall, we want to highlight the importance of functional, experimental studies with multiple omics’ biological design. Like other experimental sciences, the final purpose of microbiome study is to find the mechanism of microbiome and the links to biological and environmental factors and to establish causality of them. Thus, an experimental and functional study is more important than an operational method. However, any experimental and functional study needs statistical methods to confirm its effect.
8.5 Summary
In this chapter we first described how the commonly clustering-based OTU methods were adopted in microbiome study and how the heuristic clustering OTU methods have been developed to advance the assignment of OTUs in microbiome study. We also described the limitations of clustering-based OTU methods and the purposes for the use of OTUs in microbiome study and discussed defining species and species-level analysis. Next, we introduced the concept shifting in bioinformatic analysis and the two single-nucleotide resolution-based OTU methods: distribution-based clustering (DBC) and Swarm2, which provided the contexts of moving beyond the OTU methods. Then, we focused on two kinds of moving beyond the OTU methods: oligotyping, the entropy-based single-nucleotide resolution non-clustering OTU method, and denoising-based methods including pyrosequencing flowgrams and Illumina denoisers. In bioinformatic analysis of microbiome data, pyrosequencing techniques were early developed, while Illumina techniques represented the current development. Thus we focused on five newly developed Illumina denoisers: (1) cluster-free filtering (CFF), (2) DADA2, (3) UNOISE2 and UNOISE3, (4) Deblur, and (5) SeekDeep. We also introduced overall comparisons of denoising-based and clustering-based methods and commented on the denoising-based methods. In the final parts of this chapter, we discussed on the movement of moving beyond OTU methods, highlighting the necessity of targeting single-base resolution, discussing the possibility of moving beyond traditional OTU methods, the issues of sub-OTU methods, the assumption of sequence similarity predicting to the ecological similarity, as well as the functional analysis and multi-omics integration. Starting with Chap. 9 through Chap. 18, we will focus on investigating statistical analysis of microbiome data. In Chap. 9, we will introduce alpha diversity.